1. 引言
肠道微生物影响人体的营养代谢与免疫功能,且微生物丰度比例失衡会导致多种疾病的产生,例如肝硬化、二型糖尿病与肥胖等,因此肠道微生物逐渐成为了判断疾病状态重要标志 [1] [2] 。近年来随着高通量测序技术的发展,相关研究通常使用来自宏基因组测序分析的肠道微生物操作分类单元Operational Taxonomic Units (OTU) [3] 的丰度数据。目前已有一些机器学习模型应用于疾病预测,它能够学习微生物数据的关联和特征,并自动适应新的数据集特征,具有较好的泛化能力 [4] ,如支持向量机Support Vector Machine (SVM)、随机森林Random Forest (RF)、神经网络等分类方法已成功应用于结直肠癌、肝硬化、二型糖尿病等疾病的预测;Krizhevsky等 [5] 提出了卷积神经网络Convolution Neural Network (CNN),Tsang等 [6] 证明该方法可捕获到微生物数据的相互作用关系;Bokulich等 [7] 针对微生物数据,使用机器学习方法对微生物数据进行有监督分类学习,并与回归模型分类效果进行比较;Lo和Marculescu等 [8] 提出了用于分类宿主表型的神经网络模型MetaNN,同时为了缓解过拟合问题,对微生物数据使用了数据扩充方法;Sharma等 [9] 提出了一种基于分类学的预测方法TaxoNN,本文将使用新提出的方法与该方法进行比较。我们针对二型糖尿病患者与肝硬化患者的微生物数据,构建了两个新的预测模型:DPNC、DPVC,考虑OTU数据高维且稀疏的特点,本研究使用了非负矩阵分解Nonnegative Matrix Factorization (NMF) [10] 和变分自动编码器Variational Auto Encoder (VAE) [18] 处理原始的OTU数据。其中Karthik等 [11] 证明NMF方法可以作为无监督学习方法,在生物计算学中执行多项任务,包括分子模式发现、类比和预测、跨平台和跨物种分析等。
2. 数据集与预处理
本文分别采用实证数据与模拟数据进行模型的训练与评估,本章将介绍数据集的来源、结构和建模的流程与架构。
数据集
实证数据包括来自两项研究的数据:二型糖尿病Type 2 Diabetes (T2D)研究和肝硬化疾病LiverCirrhosis (Cirr)研究。T2D数据源自Qin 等人的研究 [12] ,共包括344个样本,其中包含174个T2D患者样本和170个对照组样本。Cirr数据取自Qin的研究 [13] ,这项研究探讨了肠道微生物组成对肝硬化疾病的影响,数据包含了118个患病样本和114个对照样本。模拟数据的生成基于克罗恩病实证研究数据,该研究由Turpin等人 [14] 提供,是Genetic,Environmental,Microbial (GEM)项目的一部分。该项目旨在跟踪监测克罗恩病患者一级亲属的身体状况,以探究克罗恩病与潜在触发因素,包括遗传学、微生物组、环境和肠道屏障等之间的关系;克罗恩病数据集共包含1796个样本,其中包括45个阳性样本。
数据预处理
本文使用的模拟数据基于克罗恩病患者和对照组的原始数据,采用数据增强方法生成了近10万个样本的模拟数据集,这一过程旨在扩大样本规模,增加数据的多样性,从而提高模型的鲁棒性和泛化能力;通过复制和引入噪声,我们能够模拟真实生物群落中存在的变异性,有助于捕捉生物数据的特征和关联。
数据增强的步骤包括对原始数据集的复制和添加白噪声。对于每个样本,我们选择了OTU变量中的非零元素并对其引入噪声。我们假设噪声数据服从高斯分布,均值范围在
,标准差为
,此外,我们使用以下公式生成疾病状态:
(1)
其中
表示第
个OUT特征或降为得到的第
个潜在特征,
表示疾病状态呈阳性,
表示疾病状态呈阴性,
为特征回归系数,
为潜在特征成对相互作用系数,
为疾病的基本流行率,本文取
,由此生成16,626个阳性样本,74,970个对照样本。
3. 模型与方法
首先选取克罗恩病患者及其一级亲属的宏基因组测序数据为例展示建模流程,使用数据增强方法生成大量模拟数据样本,使用(1)式生成疾病状态,从模拟数据中抽样获取疾病组与对照组,对OTU数据降维处理并输入分类模型,整体流程如图1所示:
3.1. 非负矩阵分解(Nonnegative Matrix Factorization, NMF)
非负矩阵分解的基本思想是将一个非负矩阵X,转换为两个非负矩阵的乘积:
(2)
为方便起见,我们称
为基矩阵,
为系数矩阵,假设
是一个
的矩阵,则
是一个
的矩阵,
为一个
的矩阵,其中
是矩阵分解所得潜在因子数量参数,矩阵
表示原始数据特征在潜在因子下的权重,矩阵
包含潜在因子在所有样本上的表达信息。
本文使用sklearn.decomposition库中的NMF函数实现矩阵分解,其中参数components决定潜在因子个数 ,通常认为当且仅当
,NMF方法实现了对原始数据的降维,图2展示潜在因子个数为8、16、32时的特征模式热图:
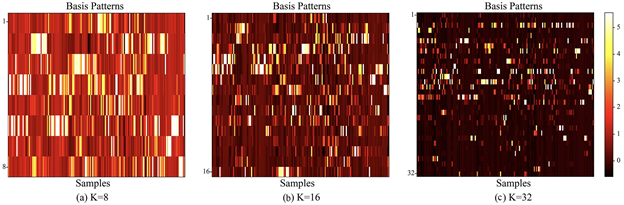
Figure 2. Heatmap of feature patterns across different latent factors
图2. 不同潜在因子下的特征模式热图
Kong等 [15] 在NMF中加入L1、L2正则化约束,以提高矩阵分解的鲁棒性,由此得到带有约束项的目标函数:
(3)
其中L1约束表示通过赋予权重,仅保留部分特征发挥作用;L2约束表示对OTU变量加以平滑约束,考虑了OTU之间的相互作用关系。Wang等人的研究发现,在植物和肿瘤基因表达数据的分析中,使用基于L1、L2范数的NMF可以提取更多特征基因 [16] ;此外,为使CNN能够更充分地捕捉到微生物数据间的空间关系、相互作用关系,本文考虑聚类变量的两种排列方法,分别是基于相关关系的排序与基于欧式距离的排序。
3.1.1. DPNCcorr
该方法首先计算属性矩阵
中特征的Spearman相关系数矩阵记为矩阵
,其中 表示NMF的聚类数目,基于下式计算聚类变量的累积相关系数:
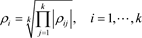 (4)
(4)
其中
表示NMF聚类得到的特征
与特征
的相关系数,式(4)计算的
则为第
个特征的累积相关系数,本文按照
值从大到小的顺序对特征进行排列,变量排序后的数据作为分类模型的输入,使得累积相关系数相近的特征能够被CNN的同一个滑动窗口捕捉到。
3.1.2. DPNCdis
需计算出NMF的聚类中心
,根据其余变量距聚类中心的距离对变量进行排序,聚类中心的计算方法如(5)式:
(5)
其中
表示NMF聚类的特征
,
表示特征
与特征
的欧氏距离,
将特征按照距聚类中心由近到远的顺序排列,并采用与
相同的方法,将排序后的变量作为分类模型的输入,使CNN的滑动窗口可以同时捕捉到。
3.2. 变分自动编码器(Variational Auto Encoder, VAE)
3.2.1. 自动编码器(Auto Encoder, AE)基本理论
自动编码器是用于处理无监督学习任务的机器学习方法,在降维和特征提取上的表现较好 [17] ,该方法分为编码层与解码层两部分,其流程框架如图3所示:
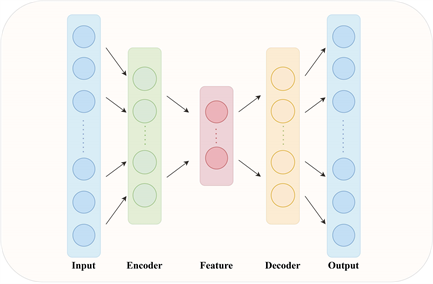
Figure 3. Auto Encoder workflow diagram
图3. 自动编码器流程图
编码器部分负责将原始数据映射到编码空间,将初始特征转换为潜在特征;而解码器则负责从编码空间中的潜在特征重构数据,尽可能还原原始输入。假设输入为
,该方法的执行过程如下:
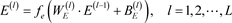 (6)
(6)
式(6)中
指编码器的层数,
和
表示编码器第
层的权重向量和偏置项向量,
即为输入
,
为低维深层特征;解码器传递公式如式(7)所示:
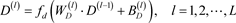 (7)
(7)
同理,
指解码器的层数,
和
表示解码器第
层的权重项和偏置项,
等同于
表示低维深层特征,
为重构数据
,自编码器的目标是最小化重构误差,即原始数据与重构数据之间的差异:
(8)
3.2.2. 变分自动编码器(VAE)基本理论
自动编码器具有映射分布无规律、空间不连续的问题,导致输入到潜在特征空间没有平滑的过度,其结果是降维得到的潜在特征矩阵稀疏,有效的潜在因子数量无法控制;实际上宏基因组变量数目庞大,相应的潜在变量空间也应连续,由Kingma [18] 等提出的VAE更适用于此背景下的数降维任务,其拟合后验分布并重采样生成数据的过程保证了潜在空间中变量连续性、生成样本多样性的要求,理论上可以优化模型过拟合情况。
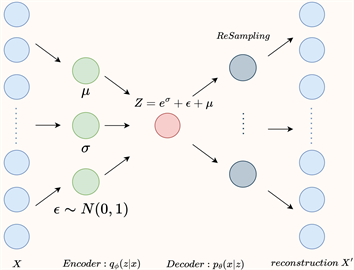
Figure 4. Variational auto encoder workflow diagram
图4. 变分自动编码器流程图
变分自动编码器的整体流程如图4所示,模型的关键思想是将数据的生成过程建模为一个概率分布,学习该分布的参数以实现降维与新样本的生成,其中模型首先通过编码器
根据给定的输入
生成潜在变量
,再通过解码器
对潜在变量
重抽样,生成新的样本。
VAE的重点在于损失函数的构造,共由重构损失与KL散度正则化项两部分组成,损失函数是二者的线性组合,分别衡量了重构样本与输入样本的相似度、潜在变量分布与先验分布之间的差异度。重构损失(Reconstruction Loss)衡量解码器
生成重构数据
与
的误差,通常使用交叉熵或均方误差:
(9)
而
的含义即在潜在变量空间中潜在变量分布
下,得到通过解码器生成重构数据分布
的对数似然最大化;另一部分则是通过KL散度衡量
与先验分布
之间的差异,即潜在分布与预先设定的理想分布之间的偏离程度,KL散度的计算如(10)式所示:
(10)
实际上我们想要得到潜在变量的后验分布
,该计算涉及对潜在变量
的计算,然而由于潜在空间维度较高的特性,使得潜在变量的后验分布的计算难以实现,因此我们使用
近似表示后验分布,
相比较以得到我们想要的分布,考虑宏基因组数据特征,我们假定
服从高斯分布;则VAE完整的损失函数为:
(11)
4. 实验结果分析
为实现分类预测效果,设置卷积神经网络的最后一层为带有SoftMax激活的两节点全连接层,其余激活函数均适用Relu;卷积层的个数为4,每个卷积层均带有一个最大池化层,卷积核个数分别为32、64、128、256;随后共有3个全连接的隐藏层,其节点个数分别为512、256、64;考虑特征数量大,可能带来训练模型过拟合的问题,我们在神经网络的全连接层中加入L2正则化处理;针对本研究的分类问题,使用预测结果绘制ROC曲线,计算对应的AUC值,比较本文提出的方法当下流行的宏基因组疾病预测方法。
4.1. 模拟数据预测结果
从模拟数据集的近10万份样本中,随机挑选200份疾病样本与200份对照样本,其中80%用作训练集、20%用作测试集;我们将提出的方法与传统的分类方法进行比较,结果如图5所示:
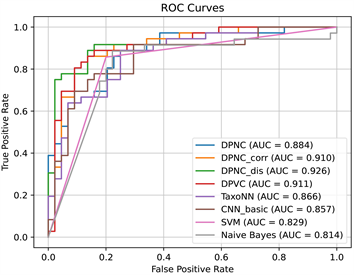
Figure 5. The ROC curves of 8 methods on the simulated dataset.
图5. 8种方法在模拟数据集上的ROC曲线
基于NMF降维的模型DPNC在模拟数据集上的预测的AUC值达到了0.884,相较于Sharma提出的TaxoNN方法(AUC = 0.866)提升了0.018;而DPNC的基于相关性与欧式距离排序的衍生模型,预测效果得到了更为显著的提升,AUC分别为0.910、0.926;本文提出的基于变分自动编码器的神经网络预测模型在模拟数据集上的表现较TaxoNN同样有显著的提升,AUC达到了0.911;而其余常见的机器学习方法如基础的卷积神经网络、支持向量、贝叶斯分类模型,预测结果所得的AUC分别为0.857、0.829、0.814。
需要说明的是,加入了变分自动编码器的DPVC的AUC值虽不是最高,但由于其具有学习原始数据分布的能力,因此一定程度缓解了训练过拟合现象,可视化训练过程如图6所示,可以看出DPVC在训练集与测试集上均有较高的预测精度,表现出更好的模型性能。
4.2. 实证研究结果
我们采用了本文提出的四种方法,将其应用于实证数据,并与TaxoNN预测方法进行比较。评估分类效果时,我们使用AUC值作为评价指标,对肝硬化数据集与二型糖尿病数据集的预测结果进行了分析:
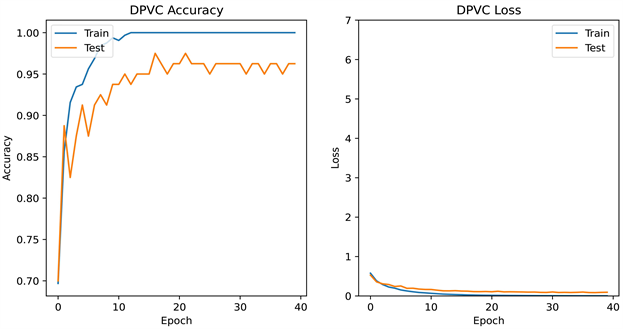
Figure 6. The performance of DPVC on the simulated dataset
图6. DPVC在模拟数据集上的表现
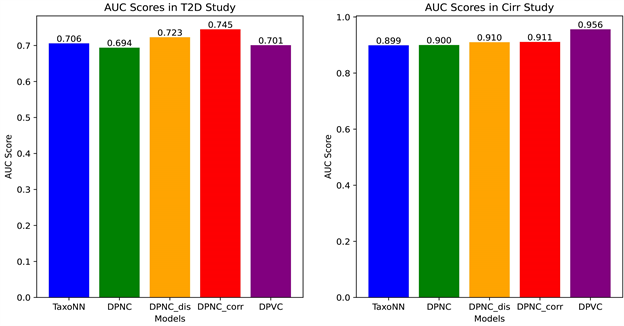
Figure 7. The performance of multiple new methods on the clinical dataset
图7. 多种新方法在临床数据集上的性能表现
参考图7,我们发现在肝硬化疾病患者预测中,加入了变分自动编码器的卷积模型DPVC表现突出,其AUC值达到了0.956。对于二型糖尿病患者的预测,基于相关性排序的
方法表现出色,其AUC值达到了0.745,明显优于其他方法。值得注意的是,在宏基因组对糖尿病发病影响研究中,仅使用VAE或NMF的两种建模方法的预测效果未超越Sharma等提出的TaxoNN,然而通过考虑对降维后特征的基于距离和相关性的重排方法,新模型的预测效果得到了显著提升,这表明该方法成功地捕捉到了微生物间存在的空间距离关系与相互作用关系。
5. 总结
本文提出了两个基于宏基因组分析的疾病预测模型。这两个模型分别利用门级别的分类学数据作为原始特征,采用非负矩阵分解和变分自动编码器方法进行数据降维。进一步,针对前者,我们引入了基于特征空间距离和相关关系的变式新模型,然后建立了带有L2正则化约束全联接层的深度卷积模型作为分类模型。
新模型在模拟数据集、肝硬化患者数据集和二型糖尿病数据集上表现出色。与之前提出的方法相比较,三个数据集上的AUC指标分别提升了0.06、0.039和0.057,显示出了显著的性能提升。