1. 引言
三疣梭子蟹(Portunus trituberculatus)是中国重要的养殖蟹类,已经吸引了广泛的研究,这需要越来越多的基因组背景知识。迄今为止,三疣梭子蟹尚没有完整的全基因组信息,而且该物种的转录组信息也很少。因此,利用下一代测序技术对其基因组和转录组进行测序分析,系统地研究三疣梭子蟹在弧菌感染下的抗性基因,不仅可以筛选到一些耐弧菌免疫基因,阐明其代谢途径,还可以挖掘到一些跟弧菌抗性相关的分子标记,为培育抗逆抗病的三疣梭子蟹新品种提供理论基础,使三疣梭子蟹健康可持续发展。
2. 前言
Sanger测序等一代测序技术曾在解析基因组序列方面发挥了巨大的作用,然而一代测序也存在着成本高、周期长、产出率低等诸多不足之处,一直无法在世界范围内得以大力推广。近些年来,随着科技进步“下一代测序”(Next-Generation Sequencing, NGS)以高通量测序(High-Throughput Sequencing)为标志,凭借其较为低廉的价格、周期短和产出率高的优势在动植物领域得到广泛的应用。目前,下一代测序的主要测序技术有美国罗氏公司(Roche)的454基因组测序仪、Illumina公司开发的Illumina测序仪和ABI公司的SOLID连接酶测序平台,这些测序平台均采用循环芯片测序法(cyclic-array sequencing),并被誉为第二代测序技术 [1] [2] 。
全基因组测序(whole genome sequencing)是对未知基因组序列的物种进行个体的基因组测序,能够快速的鉴定到大量高密度的SNP位点,可用于重要候选基因的筛选、遗传变异检测及群体遗传进化分析等,因而全基因组测序广泛应用于群体进化、群体结构、种群历史、遗传定位和连锁图谱的构建,例如王金昌等对海洋贝莱斯芽胞杆菌Bam-6基因族进行注释发现了贝莱斯芽胞杆菌代谢合成物的同源物、毛明光等对太平洋鳕鱼线粒体基因族进行测序分析发现一段保守序列 [3] [4] [5] 。WGS研究主要包括两方面,一方面为全基因组从头测序(de novo),另一方面为全基因组重测序(re-sequencing) [6] 。
重测序是以物种的参考基因组序列为依据,进行个体或群体间的基因组测序,并对其差异信息进行分析的一种测序方法。相比较于传统的方法,重测序作为二代测序具有许多优点:1) 信息全面,可以获得全基因组的序列信息;2) 信息精确,可以精确挖掘到每个SNP位点,直接找到致病位点;3) 产出效率高,可以挖掘到许多性状相关的关键基因 [7] 。通过全基因组重测序技术,可以获取到大量的遗传变异信息,实现DNA分子水平上的遗传分析并筛选到性状相关的候选基因。
关于哺乳动物的研究中,利用WGS技术进行遗传分析已经得到广泛应用。Leif Andresson团队对9个群体的鸡进行WGS测序筛选到3个重要的驯化基因;黄路生教授团队对11个中国地方猪种和3个野生猪种进行了WGS测序,筛选到了210个与环境适应性相关的基因;Stothard团队运用重测序的方法首次在美国荷斯坦牛和黑安格斯牛上开展了拷贝数变异(copy number variation, CNV)检测 [8] [9] 。尽管重测序在哺乳动物中已经取得了许多成绩,但在水产动物中仍少见报道。
3. 材料和方法
3.1. 实验材料
实验材料均来自于青岛黄海水产研究所,分别来自于健康存活未感染副溶血弧菌的80日龄梭子蟹和感染副溶血弧菌72 h后存活梭子蟹的肌肉组织。
3.2. 建库、测序及分析
将检验合格的DNA样品等量混合为两个混合池,分别命名为易感DNA混合池(CG)和耐感DNA混合池(CT) [10] 。通过Covaris破碎机随机打断混合DNA样品成350 bp片段,使用Truseq Library Construction Kit对其建库,建库过程中严格使用说明书中推荐的试剂和耗材。350 bp片段通过终端修复、ploya尾、测序接头、纯化、PCR扩展等一系列步骤后,整个文库就制备完成。
建库具体流程如图1所示。文库制备完成后,采用Qubit2.0进行初步定量,将文库稀释至1 ng/ul。然后使用Agilent 2100检测文库的insert size。待检测insert size符合标准后,为确保文库质量,还需采用Q-PCR法准确定量文库有效浓度(文库有效浓度 > 2 nM)。文库浓度检测合格后,再按有效浓度和目标下机数据量的需求pooling对不同文库进行Illumina HiSeq TM PE150测序 [11] 。
由于测序获得的是raw reads或 Sequenced Reads,其中可能带有大量低质量的reads,为了得到clean reads,需要对raw reads进行信息质量分析,其步骤如下 [12] :
1) 去除带接头(adapter)的reads pair;
2) 当单端测序read中含有的N的含量超过该条read长度比例的10%时,需要去除此对paired reads;
3) 当单端测序read中含有的低质量(Q ≤ 5)碱基数超过该条read长度比例的50%时,需要去除此对paired reads。
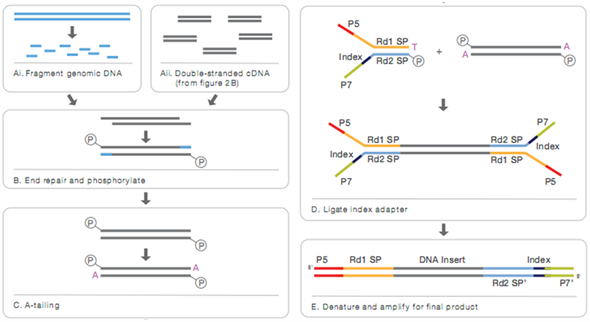
Figure 1. Library construction sequencing process
图1. 建库测序流程
应使用Burrows-Wheeler alignment tool (BWA)比对软件将过滤后的有效数据与参考基因组进行比对,参考基因组统计信息见下表1所示,使用SAMTOOLS软件去除比对结果重复 [10] 。获得的数据可以用于后续注释分析及SNP/InDel检测,可实现DNA水平差异功能基因注释及差异基因挖掘,全基因组重测序的生物信息分析流程如图2所示。
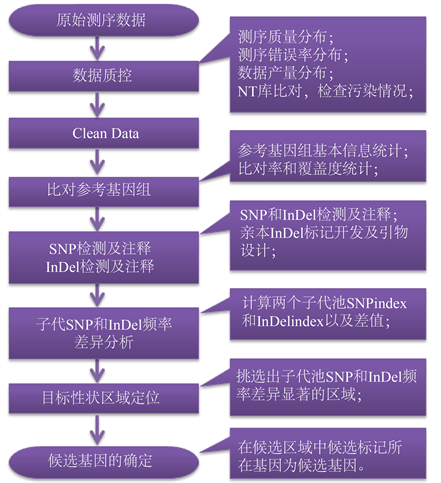
Figure 2. Whole-genome biological information analysis process
图2. 全基因组生物信息分析流程

Table 1. Statistics of reference genome
表1. 参考基因组基本情况统计
3.3. QTL-seq分析
3.3.1. SNP/InDel检测和注释
SNP (single nucleotide polymorphism,单核苷酸多态性)是指在基因组水平上由于单核苷酸发生变异而引起DNA序列产生多态性,包括单碱基的颠换或转换等 [9] 。而InDel则是表示基因组产生小片段的缺失和插入序列。我们采用Genome analysis toolkit 3.8 (GATK)软件中的UnifiedGenotyper模块检测SNP和InDel,SNP过滤参数设置为:MQ < 40,QD < 4,FS > 60;InDel过滤参数设置为QD < 4,FS > 200 [7] 。
3.3.2. SNP频率差异分析
我们以参考基因组作为参考,分析计算易感和耐感个体在每个SNP位点中的SNP-index (SNP的频率)。如图3所示是对个体池中SNP-index计算的一种统计方法,其原理是以参考基因组或某一亲本作为参考,通过测序reads以便于对碱基位点的碱基进行统计分析。统计在某一个碱基位点处个体池和亲本或者参考基因组是否出现不同或相同的reads,并统计其中不相同条数占总条数的比例,该比例即为SNP-index。其中,完全与其参考基因组或亲本完全与其不同的SNP-index记为1,相同的则记为0。按照此方法计算出敏感池和耐感池的全部SNP-index。为减少测序错误和比对错误造成的影响,对计算出SNP-index后的多态性位点进行过滤,过滤标准如下:
1) 两个个体中SNP-index都小于0.3,并且SNP深度都小于7的位点,过滤掉;
2) 一个个体SNP-index缺失的位点,过滤掉。

Figure 3. SNP-index calculation method
图3. SNP-index计算方法
得到过滤后的多态性位点后,对SNP-index在染色体上的分布进行作图。默认选择1 Mb为窗口,1 kb为步长,计算每个窗口中SNP-index的平均值来反应个体的SNP-index分布。同时对SNP频率差异分布进行作图,计算Δ(SNP-index),即两个个体SNP-index作差:Δ(SNP-index) = SNP-index (耐副溶血弧菌弧菌性状B) – SNP-index (副溶血弧菌敏感性状A)。进行1000次置换检验,选取95% (蓝色)置信水平作为筛选的阈值。
3.3.3. InDel频率差异分析
InDel-index分析方法同1.3.2。
3.4. 候选标记挖掘
3.4.1. 候选标记筛选
为了不忽略掉微效QTL的影响,在全基因组范围内挑选候选SNP和InDel,如果参考亲本和子代表型相同,则挑选子代池中All-index接近0的位点;如果参考亲本和子代表型相反,则挑选子代池中All-index接近1的位点作为候选位点。
SNP标记筛选原则:
1) 选择个体间Δ(SNP-index)接近1的标记;
2) 选择多个标记位于一个Conting上的;
3) 对产生移码突变或stop gain或stop loss或非同义突变或者可变剪接位点的位点优先进行筛选。
InDel标记筛选原则:
1) 选择个体间Δ(InDel-index)接近1的标记;
2) 选择多个标记位于一个Conting上的;
3) 按照插入片段长度进行排序。
引物设计原则:
1) 避开在标记左右50 bp处设计引物;
2) 扩增片段在300~500 bp左右;
3) 引物长度在18~23 bp;
4) 引物退火温度控制在55℃~65℃之间,上下游引物温度差最好控制在3℃之间;
5) 引物GC含量控制在40%~60%之间,上下游引物GC含量差最好不要超过5%。
3.4.2. 耐副溶血弧菌标记的验证
采用PCR产物测序的方法在CG和CT群体里对耐副溶血弧菌性状相关候选分子标记进行验证:
1) 首先在标记位点侧翼序列设计引物,其中至少有一条引物距离标记位点70 bp以上;
2) 利用设计好的引物分别以CG和CT混合DNA材料为模板进行PCR扩增,并将成功扩增的PCR产物进行测序,测序引物选择离标记位点较远的引物;
3) 利用ContigExpress软件分析测序峰图,挑选CG和CT两组在对应位置测序峰图有较大差异的标记继续进行个体DNA模板的PCR扩增和测序分析;
4) 根据测序结果统计每个个体的基因型,并通过SPSS软件分析标记与耐副溶血弧菌性状是否相关 [13] 。
具体的操作步骤如下:
1) PCR扩增体系和程序
利用全式金公司的高保真酶进行PCR扩增。设置PCR反应程序为1,95℃,2 min;2,95℃,20 s;3,55℃,20 s;4,72℃,30 s;2~4,35个循环;5,72℃,5 min;6,4℃保存。PCR反应体系见图4。
2) 电泳检测
用琼脂糖配置1%的电泳胶,把制好的琼脂糖凝胶放入水平电泳槽中,进行电泳检测实验,电泳时间设定为30 min,电泳结束后用凝胶成像系统观察并拍照记录,切割出明亮且条带单一的电泳胶样品送置青岛擎科生物有限公司进行测序。
3) 统计分析
利用ContigExpress软件对测序峰图进行分析,选择耐感群体混合模板和易感群体混合模板中碱基位置出现显著差异的引物,并继续使用该引物,以每个个体中进行PCR扩增验证,扩增条件和上述一致,然后将明亮且位置大小符合的电泳条带送去测序 [10] 。
再将返回的个体测序结果用ContigExpress软件进行观察并将基因型信息导入SPSS软件,利用卡方检验计算P值,选出P < 0.05的标记为候选标记。
4. 结果与分析
4.1. 全基因组重测序结果
由于现阶段的技术不足,需要在测序前添加上一些接头,导致部分测序结果中也会含有冗余的接头序列信息;此外,测序时也可能会产生一些低质量的序列信息;因此对序列的质量进行评估以及过滤对后续的结果分析极为重要。经测序质量分布检查、测序错误率分布检查以及测序数据过滤后,统计结果表明测序结果极佳,质量都在Q30以上,错误率低,有效数据质量高,如图5和图6所示。
本次测序共产生Raw data高达51.963G,过滤后的Clean data也有51.867G,本次测序质量高(Q20 ≥ 94%、Q30 ≥ 87%),GC含量也在41%左右。因此,本次实验样本的数据量充足,GC分布正常且测序质量高,符合建库测序成功标准。测序质量数据汇总见表2。
使用BWA软件将易感池和耐感池的测序数据和梭子蟹参考基因组进行比对。比对结果表明,所有样本的比对率在85%以上,对参考基因组(排除N区)的平均覆盖深度在25X以上,1X覆盖度(至少有一个碱基的覆盖)在79%以上。比对结果正常,可用于后续的标记检测分析。具体Reads与参考基因组比对情况统计如表3所示。
Table 2. Summary of the quality of sequencing data
表2. 测序数据质量情况汇总
Table 3. Sequencing depth and coverage statistics
表3. 测序深度及覆盖度统计
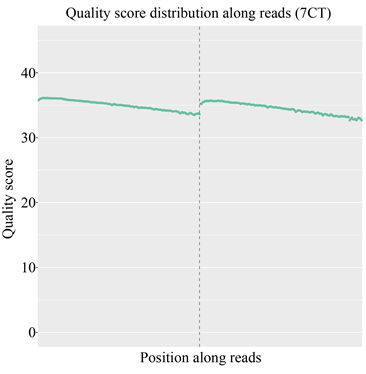
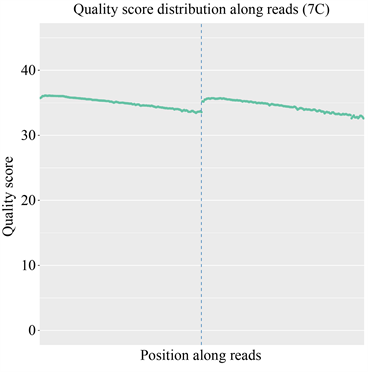
纵坐标为单碱基错误率,横坐标为reads的碱基位置;前150 bp为双端测序序列的第一端测序Reads的质量值分布情况,后150 bp为另一端测序reads的质量值分布情况。
The abscissa is the base position of reads and the ordinate is the single base error rate; the first 150 bp is the quality value distribution of the first-end sequencing reads of the double-end sequencing sequence, and the last 150 bp is the quality value distribution of the sequencing reads at the other end.
图5. 测序质量分布图
4.2. SNP/InDel检测与注释
有效数据经与参考基因组比对,分别检测到36,929个SNP标记和145,790个InDel位点。候选位点的注释情况如表4和表5所示。
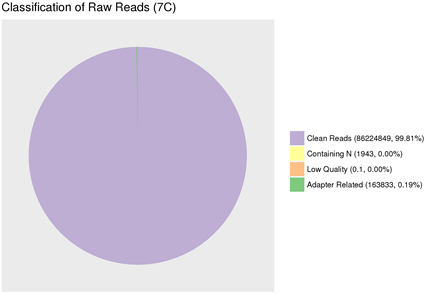
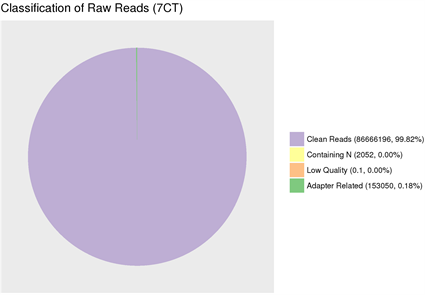
(1) Adapter related:因有接头,过滤掉的 reads数及其占总raw reads数的比例。(2) Containing N:因N含量超过10%,过滤掉的reads数及其占总raw reads数的比例。(3) Low quality:因低质量,过滤掉的reads数及其占总raw reads数的比例。(4) Clean reads:最终得到的clean reads数及其占总raw reads数的比例。
(1) Adapter related: Due to the adapter, the number of reads filtered out and its proportion to the total number of raw reads. (2) Containing N: because the N content exceeds 10%, the number of reads filtered out and its proportion to the total number of raw reads. (3) Low quality: due to low quality, the number of reads filtered out and its proportion to the total number of raw reads. (4) Clean reads: The final number of clean reads and their proportion to the total number of raw reads.
图6. 原始数据过滤结果
Table 4. Statistics of SNP detection and annotation
表4. SNP检测及注释结果统计
Table 5. InDel detection and annotation result statistics
表5. InDel检测及注释结果统计
4.3. 重测序SNP/InDel位点验证分析
经SNP和InDel合并后的频率差异分析后,得到成功注释的SNP标记257个,InDel标记187个。在注释结果中选择了55个SNP标记和32个InDel标记进行验证,初步验证结果表明23个SNP标记中有10个标记与检测结果一致,阳性率为41.82%;32个InDel标记中有10个标记与检测结果一致,阳性率为31.25% (InDel标记由于序列的复杂性导致测序结果较低,可能对阳性率造成了影响)。
继续利用上述位点存在显著差异的引物,分别在易感个体和耐感个体上进行扩增测序,最终筛选出9个SNP标记和2个InDel标记,统计结果如表6所示。
Table 6. Markers related to Vibrio resistance and prediction of their gene functions
表6. 与弧菌抗性相关标记及其定位基因功能预测
5. 讨论
由于基因组学研究技术手段的飞快发展,使得检测大规模、高通量的动物基因组内的变异位点变得越来越容易 [14] 。先前的研究报道表明,不同物种的不同种群SNP频率不同,而有关三疣梭子蟹抗弧菌反应相关的免疫相关SNP信息鲜有报道 [15] 。因此,对三疣梭子蟹抗病抗逆相关基因及其SNPs进行研究,有助于提高对三疣梭子蟹免疫防御机制的认识,有利于梭子蟹抗病品种的筛选和培养。为得到高质量的检测数据及结果,本研究使用了一种基于BSA混合池的全基因组重测序技术对对三疣梭子蟹易感群体和耐感群体之间可能存在的抗病相关的差异基因进行了深入挖掘。相比于传统的抗病QTL的定位方法,本研究所用的全基因重测序技术不仅省时省力,并且可以从分子层面直接挖掘与抗病相关的基因。
目前Illumina公司的测序平台是应用最为广泛的二代测序平台,本研究即采用Illumina HiSeqTM PE150平台进行测序。碱基的质量高低与测序错误率息息相关,鉴于当前测序技术扔存在局限性,因此测序片段前段和末端几个cycles的错误率会偏高 [16] 。测序获得的碱基质量值用一般Qphred值表示,如果测序错误率用e表示,则Qphred = −10log10(e),Q10、Q20、Q30和Q40表示不正确的碱基识别分别1/10、1/100、1/1000和1/10000,即碱基正确识别率分别为90%、99%、99.9%和99.99%;根据重测序的结果表明,本研究共产生51.983G的数据,Q20平均值 ≥ 94.23%,Q30平均值 ≥ 87.54%,测序质量较高。重测序需要将产生的Clean reads通过BWA软件比对到梭子蟹的参考基因组上。结果表明,两个群体的所有样本跟梭子蟹参考基因组的比对率在85%以上,与参考基因组具有较高的相似性;同时,所有样本对参考基因组的平均覆盖深度在25X以上,1X覆盖度在79.08%以上,4X覆盖度在76.69%以上,这些结果都表明该测序数据的均一性和参考基因组序列的同源性较高,数据质量高。
SNPs是基因组中广泛存在的突变,基于基因组中的不同位置,SNP可以通过不同的机制来影响基因的翻译或转录,基因调控区内SNP发生突变可能会影响相关基因的表达像周慧等发现miR-17-92基因启动子区rs1813389 A/G碱基发生颠换可能于子宫内膜癌症有关;非同义编码区SNP突变直接改变基因编码蛋白的氨基酸组成,对蛋白质功能域的出现具有至关重要的作用像黎江溪发现TNNC1基因编码区第44个碱基由G转换成C可能造成肥厚型心肌病高风险 [17] [18] [19] 。先前的研究主要集中在非同义突变SNP (nonsynonymous single nucleotide polymorphism, nsSNP),因为这些SNP最有可能直接影响蛋白质的结构和活性;然而近些年来,越来越多的研究表明,内含子和基因间的SNPs也可能在性状的变化中起着决定性作用 [20] 。例如,F-box和富含亮氨酸重复蛋白17 (leucine rich repeat protein 17, FBXL17)基因的第三内含子中的SNP突变解释了拌花生和他性别逆转中存在了58.4%的表型变异 [21] 。因此,我们在测序时对三疣梭子蟹内含子和基因之间区域SNPs进行分析。
6. 小结
在本研究中,从两个群体的WGS分析中共鉴定出257个SNP和184个InDel,并成功进行了功能注释,并没有检测到非同义突变,大部分标记处于内含子和基因之间。其中位于内含子区域的SNP和InDel分别占比18.29%和25%;位于基因之间区域的SNP和InDel分别占比78.6%和71.7%。