1. 引言
近些年来,单细胞RNA测序技术在众多领域都有着重大进展,已然成为了研究细胞动力学的一种强有力的工具。单细胞RNA测序技术不但能够帮助基因学者更好地探索单细胞水平的基因表达谱,了解在不同组织微环境下细胞表达及功能差异,也为发现罕有细胞、细胞谱系分化轨迹提供了前所未有的机会。目前,单细胞RNA测序已成为研究细胞异质性的关键生物学问题的首选。该类技术的进步为单细胞RNA数据分析提供大量数据的同时,也在基因数据分析方向上提出了挑战,其中包括如何根据测序信息进行准确的细胞分类,并找到细胞差异化表达的基因等一系列问题。
在单细胞RNA测序数据的细胞聚类问题上,Kiselev,Kirschner,Schaub [1] 等人提出了一种具有一致性的单细胞聚类算法SC3,该方法先对基因数据应用主成分分析法将基因维度降到总基因的4%到7%,然后对所有主成分进行k-means聚类,最后对所有结果进行一致性聚类,经对比该聚类算法具有良好的稳定性;而Guo,Hui,Steven [2] 等人提出了一种新的单细胞RNA序列分析流程SINCERA,在该分析过程中运用了无监督的分层聚类,并运用t检验和Wilcoxon秩和检验筛选出差异性表达基因;Yang,Liu,Lu [3] 等人提出了一种迭代聚类算法SAIC,该聚类算法需要提前选取聚类的数目和阈值,然后再进行聚类,并通过方差分析选出显著差异性表达基因。
伴随着机器学习技术的飞速发展,包括主成分分析在内的一些机器学习方法广泛地应用于基因数据分析领域。以常用的线性主成分分析方法为例,在对细胞基因数据分析时,同类细胞会被同一主成分解释,而不同主成分间的线性无关则代表着细胞间的部分差异。但基因表达数据具有强稀疏的特殊性,也即基因的维度远远大于细胞的维度,这与线性主成分样本数据服从高斯分布的前提不符,使得上述方法在单细胞RNA测序数据降维聚类的问题上受到限制。因此需要选择非线性降维方法来对单细胞RNA测序数据进行降维聚类分析。
本文应用t-SNE和UMAP两种非线性降维聚类方法,对单细胞RNA测序数据进行降维聚类分析,并将聚类结果与线性主成分分析方法降维聚类的结果进行对比,通过比较聚类图中细胞类内、类间的距离属性得出UMAP方法降维聚类效果更为理想,进而在其聚类结果的基础上筛选出显著差异化表达基因。
2. 基于t-SNE与UMAP的降维方法
2.1. t-SNE降维
2.1.1. 随机邻近嵌入(Stochastic Neighbor Embedding, SNE)
在介绍t分布–随机邻近嵌入(t-distributed Stochastic Neighbor Embedding, t-SNE)方法之前,首先介绍随机邻近嵌入(Stochastic Neighbor Embedding, SNE)方法。SNE首先将数据点间的高维欧式距离转换成了代表着相似度的条件概率 [4]。将数据点
到数据点
的相似度用条件概率
来表示,也就是说
将会以概率
挑选
为其临近点。那么在以
为中心点的Gauss分布下,可以定义
与
的相似度为:
(1)
其中
是以
为中心点的Gauss分布的方差,
代表欧式范数。如果数据点距离越近,那么
越大;如果数据点距离越远,那么
越趋近于无穷小。
对于和高维空间中的点
和
相对应的在低维空间中的点
和
,同样可得点
到点
的相似度
,在Gauss分布下,方差为常数值
时,
(2)
显然,如果点
到点
可以完全模拟高维数据点
到数据点
的相似度,那么相应的条件概率
和
应该完全相同。若考虑高维空间下所有点与
的相似度,则可构成一个条件概率分布
,同理在低维空间存在一个条件概率分布
且应该与
一致。由此可选用Kullback-Leibler距离(KL距离)来衡量两个分布的相似性,构造(3)式代价函数
,运用梯度下降的方法来使
达到最小来确定低维空间,具体形式如下:
(3)
其梯度为:
(4)
2.1.2. t-分布随机邻近嵌入(t-distributed Stochastic Neighbor Embedding, t-SNE)
虽然SNE也可以对数据进行降维,但是SNE注重的是局部结构而非全局结构。由于t分布是重尾分布,对异常点不是十分敏感,所以在对称SNE的基础上将低维空间中的分布选取为t分布。这样一来,在降维过程中,t分布的处理会使同一簇中的数据更加紧密,不同簇之间的数据更加稀疏,效果显著提高。自由度为1的t分布重新定义的
形式如下:
(5)
相应的梯度形式为:
(6)
2.2. UMAP降维
均匀流行逼近和投影(Uniform Manifold Approximation and Projection, UMAP)是一种基于黎曼几何和代数拓朴的理论框架构建的流行学习算法。它依据高维空间映射到低维空间相似度的定性结论,将高维数据的拓扑结构进行低维映射以达到降维结果 [5]。
在UMAP降维过程中,设高维数据点
,低维数据点
。令
为其度量空间,给定一个超参数
,可以得到
在
下的近邻集合
。那么高维模糊拓扑结构可使用指数概率分布表示如下:
(7)
其中,
是点
到首个最近邻数据点的距离。
是
最近邻数据点的直径。需要注意的是,这并不是一个对称函数,所以应该将该函数对称化:
(8)
在高维分布中建立模糊拓扑后,需要在低维分布中构建概率分布:
(9)
其中超参数
[6]。
UMAP希望相关数据点在投影空间中尽可能地靠近,而不相关数据点尽可能地远离。因此,引入如下函数:
(10)
在上式中,
是高维分布中数据点的权重,
是低维分布中数据点的权重。算法首先对数据集中相似的数据点施加Attractive (引力),并对非相似的数据点施加Repulsive (斥力),接着通过模拟退火优化算法逐渐减小Attractive和Repulsive,最终达到收敛。
3. 实例分析
在进行正式的降维聚类工作之前,我们对单细胞RNA测序数据 [7] 进行前期的预处理工作,在描述性分析工作中发现该基因数据具有较强的稀疏性,又根据坏死细胞和破损细胞的基因表现特征对随细胞进行筛选和质量控制,最后对数据进行标准化处理,得到基因种类8315个、细胞个数750个的单细胞测序数据。接下来对处理过后的基因数据进行降维聚类分析。
3.1. 降维聚类结果
为比较t-SNE与UMAP两种非线性降维聚类方法在基因测序数据应用的优越性,我们先对预处理后的单细胞RNA测序数据进行线性主成分降维聚类,聚类结果见图1。
而后我们对预处理后的单细胞RNA数据依UMAP及t-SNE两种非线性方法进行降维聚类 [8],细胞聚类结果见图2。
观察上文中两细胞聚类散点图我们可以发现三种降维聚类方法都将细胞分为0~4共五个类别,但从聚类效果来看,图1中线性主成分降维聚类结果类内点位距离较大、类间点位距离较小、类边缘界限模糊不清,且不同细胞类别之间有不同程度的位置重合。图2中左图为应用UMAP方法后的降维聚类的结果,右图为应用t-SNE方法后降维聚类的结果,通过对比可以明显的观察到UMAP方法的聚类结果中不同细胞类别类间距离更大,类内距离更小,且细胞点位分布更密集;而相较于UMAP聚类结果,t-SNE方法的聚类结果中类间细胞点位距离较小、类内细胞点位距离较大且细胞点位分布较为分散。综上所述,我们认为非线性UMAP降维聚类的效果最为理想。
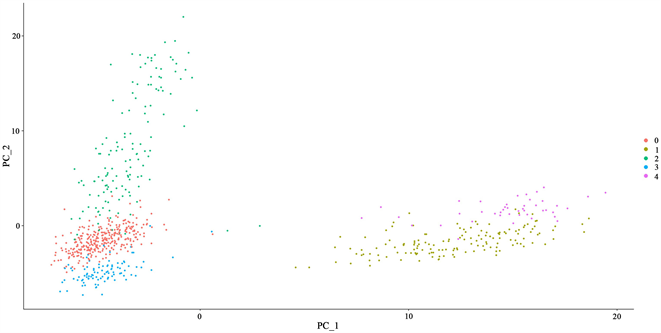
Figure 1. Cell scatter plot based on linear principal component dimensionality reduction (PCA) clustering
图1. 基于线性主成分降维(PCA)聚类的细胞散点图
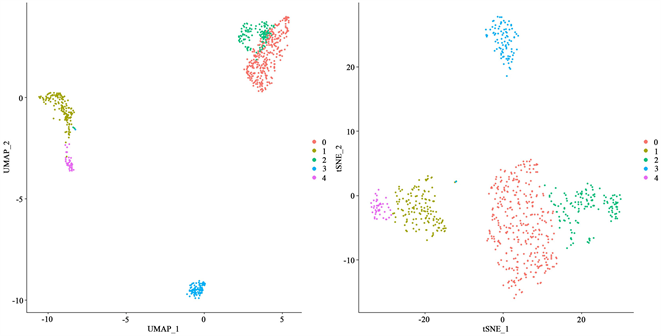
Figure 2. Cell scatter plot based on UMAP and t-SNE nonlinear dimensionality reduction clustering
图2. 基于UMAP和t-SNE非线性降维聚类的细胞散点图
3.2. 显著差异性基因筛选结果及分析
细胞分类的不同往往可能源于其显著表达基因的差异化,UMAP非线性降维方法的聚类结果能有效帮助探索识别具有显著差异性的基因。我们计算基因在不同细胞类别表达的细胞比例及其差值、基因在不同类别之间表达量差异的倍数,并依照假设:
H0:基因在两组别之间没有显著性差异;
H1:基因在两组数据之间有显著性差异。
进行Wilcoxon秩和检验,在此结果上进一步筛选出各类别的显著差异化基因。
以类别0的显著差异化基因为例生成部分差异基因列表(见表1),其中基因在类别0的细胞内与其他类别细胞内的含量差值(Difference)为降序排列,也即基因表达的差异化依表顺序依次递减。
表1中各基因的avg_log2FC值均为正值,说明这些基因在类别0的细胞群体内平均表达的折叠度高于其他类别的细胞,且由Wilcoxon秩和检验得出的p_val值远小于
的临界值,也即拒绝基因在两组别之间没有显著差异的原假设。基因在0类别细胞内与其他类别细胞内的含量差值(Difference)均为正值,这也进一步佐证表中基因为0类别细胞区别于其他类别细胞的显著差异性基因。

Table 1. Some significantly different genes with tag category 0
表1. 部分标签类别为0的显著差异性基因
注:gene:基因名称;FC (fold change):组间基因表达量的差异倍数;avg_logFC:基因在组间的平均表达量取log2,正值表明该基因在当前组中表达更高;pct.1:在当前类别细胞中检测到该基因表达的细胞比例;pct.2:在其它类别细胞中检测到该基因表达的最大细胞比例;p_val:Wilcoxon秩和检验所得p值;Difference:pct.1 − pct.2 [9]。
因此,我们分别取每组表达最高且与其他类别细胞之间含量的差值最大的前五个基因作为显著差异性基因的结果,它们分别是CD3D、IL7R、LDHB、IL32、CD3E (类别0);S100A8、LGALS2、FCN1、S100A9、LST1 (类别1);NKG7、CST7、GZMA、CCL5、CTSW (类别2);CD79A、CD79B、MS4A1、TCL1A、LINC00926 (类别3);FCGR3A、IFITM3、CD68、CFD、CFP (类别4)。
3.3. 基于基因分布热图、分布点图的差异性基因验证
为了以更加直观的方式展示上述各基因的显著差异性,我们绘制了部分含有不同显著差异性基因的细胞分布热图和部分显著差异性基因的分布点图。
我们选择每组显著差异性基因的前两个共计10个基因绘制分布热图,如图3所示,其中图例中颜色深浅为表示基因分布的密度标识:数值越大、颜色越深,说明差异性基因在对应分类区域中密度越大。从图中我们可以清晰地观察到,每个基因在细胞聚类图中的分布都较为集中,说明该基因能够较为准确的代表所属聚类区域细胞的基因特征属性。
而以IL7R、S100A8、NKG7、CD79A、FCGR3A这五个显著差异基因为例,观察其基因分布点图,如图4所示,坐标轴的横坐标为显著差异性基因名称,纵坐标为细胞类别,其中点的大小代表含有该基因的细胞比例,颜色深浅代表平均表达水平,通过比较图中分布点的颜色和大小,我们可以清楚地判断上述基因在各自不同的细胞类别内有着较高的表达水平,且表达程度远远大于其他组别。这也进一步说明筛选出的差异化基因在细胞分类的过程中起到了显著作用。

Figure 3. Heat map of significantly different gene distribution in different groups
图3. 不同组别下显著差异性基因分布热图
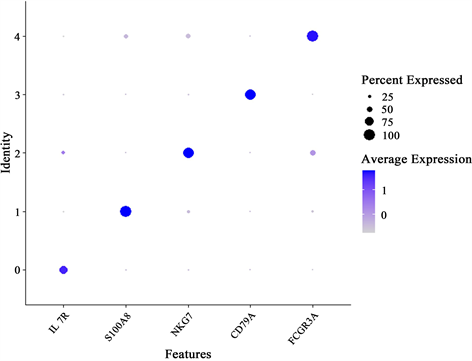
Figure 4. Dot plot of the distribution of significantly different genes in different classes
图4. 显著差异性基因在不同类别中的分布点图
4. 模型评价
通过对上文中单细胞RNA测序数据降维聚类的结果进行对比和对不同降维聚类模型特点进行归纳总结,我们得出以下结论:
线性主成分降维聚类方法受线性模型的限制,在特征选择的过程中没有进行基因筛选,降维后的基因数据对细胞分类的解释能力相对减弱,且细胞分类的区分效果在基因投影之后有所削减,反而可能使部分细胞混杂在一起难以准确区分 [10]。
不同于线性主成分降维聚类的方法,t-SNE和UMAP可以直接将高维空间的结构特征投影到低维空间中,通俗地讲,就是用平面或立体空间内点的疏密远近表现其在原本多维度状态下的疏密远近,这样能够更大程度上保留细胞原有的基因的差异化特征。而UMAP又是一种非线性流形学习算法,相比于线性主成分降维方法只适用于高斯分布样本的限制,UMAP在非高斯大容量样本上有着良好的性能。在数据降维过程中,UMAP可以保留样本全局结构,最大程度上维持样本数据完整性,并且不需要人工去选择核函数,规避了人工选择核函数对其降维性能的影响,因此,UMAP比t-SNE和线性主成分降维方法有更强的工程适用性。
基金项目
国家自然科学基金面上项目:超高维复杂数据统计降维研究(11771215),2018.1~2021.12。