1. 引言
癌症是一类由于细胞分裂和凋亡机制失常而导致的疾病,通常表现为恶性肿瘤 [1]。由于其早期诊断困难,复发率和死亡率高,目前已经成为严重威胁人类健康的疾病之一。根据国际癌症研究中心(IARC)的报告,2015年,全球约有1410万例新增癌症病例,死亡人数达到820万。据报告估计,20年后,每年新发癌症病例将增至2200万例,与此同时,癌症致死人数将从每年约820万人增至每年1300万人。而在我国,每年因癌症而死亡的人数高达280万,占全球的34.1%,且呈现出明显的上升趋势。因此构建合适的疾病诊断模型对于癌症的预防和治疗具有重要的实际意义。
传统上,利用各种类型的外科活组织检查(如骨髓活检或针活检)对癌症进行诊断。然而,由于手术的侵入性和肿瘤活检的潜在取样偏差,手术活检往往不是首选 [2]。作为替代方法,研究人员和临床医生一直在寻找用于疾病诊断的分子生物标记物。目前的诊断系统都是针对肿瘤组织的基因表达数据进行的,具体做法为:将疾病样本和正常样本进行直接比对,将比对出来的差异表达基因作为预测因子,利用机器学习中的分类算法进行构建。然而,肿瘤组织是高度异质的复杂组织,除癌症细胞外,它还包含一定数量的其它细胞,如正常细胞、免疫细胞、基质细胞、血管细胞等 [3],因此其通过高通量实验所得到的基因组/表观组数据是其所包含细胞类型信号的加权平均值。由于不同的细胞类型具有不同的分子表达谱,并且细胞组成比例在不同的组织样本间也会发生一定的变化。当在不同生物学条件下观察到的组织样本间的差异实际是由于细胞组成比例的变化引起时,这可能导致错误或误导性的发现 [4]。乳腺癌数据分析发现,利用此方法进行疾病预测准确度仅达到80% [5]。
为解决上述问题,本文从肿瘤组织的DNA甲基化芯片数据出发,在正态分布假设下构建了肿瘤样本甲基化水平的混合概率模型,利用异质性分解算法Tsisal [6] 识别疾病和正常之间的真实变化信息,将识别出的细胞类型组成比例和细胞类型特异性的甲基化位点作为生物标记物,利用SVM算法构建疾病预测模型,并通过TCGA两种癌症类型数据分析来说明所提模型的优越性。
2. 基于DNA甲基化芯片数据的疾病预测模型构建
模型的训练数据是临床某肿瘤组织样本的450 K甲基化芯片数据Y和N名受试者的疾病状态Z,其中N名受试者中包含N0个病人和N1个健康人。Y是M × N的矩阵,M 是CpG位点的个数。Z是长度为N的二进制向量(1表示疾病状态,0表示健康状态)。本文的目标是在训练数据上构建疾病预测模型使用矩阵Y来预测疾病状态Z,并对模型进行评价,继而对未知疾病状态的临床样本甲基化数据进行预测分析。
疾病预测模型构建包含三个步骤:细胞类型特异性甲基化位点识别、利用异质性分解算法估计细胞类型组成比例、利用SVM算法进行疾病分类预测。具体实施方案如下:
细胞类型特异性的位点识别是疾病预测模型构建的第一步,本文采取方差分析(选取方差最大的1000个位点)、变异系数(选取变异系数最大的1000个位点)、几何算法SISAL (每一个细胞类型特异性排名前50的位点)来识别细胞类型特异性的特征。
第二步实施过程中由于肿瘤组织是高度异质的复杂组织,其甲基化矩阵 是所包含细胞类型甲基化谱矩阵W与细胞类型所占比例矩阵H的乘积,即
。从该式子可知,疾病和正常组之间在W或H上的差异将会导致
之间的差异。因此,有必要开发反卷积分解算法去识别疾病和正常样本之间真正的变化信息。对矩阵Y利用各种分解算法求解H的过程叫做反卷积,反卷积的目标是求解出比例矩阵H,该矩阵是第三步SVM的输入。目前常用的反卷积算法主要分为两大类:基于参考基矩阵的方法(reference-based, RB) [7] [8] 和不依赖参考基矩阵的方法(reference-free, RF) [9] [10]。RB方法要求参考基矩阵W已知,组份矩阵H通过回归模型进行求解。RF方法不需要参考数据,由于W和H矩阵均为非负矩阵,因此该方法为经典的非负矩阵分解问题。相对于RB方法,RF方法需要的先验信息少,求解方法也较为成熟,因此有更广泛的应用空间。本文采用2021年张伟伟等人 [6] 开发的RF反卷积算法Tsisal进行求解比例矩阵H,它从肿瘤组织的甲基化数据出发,利用几何算法SISAL求解甲基化数据构造的单纯形,估计得到细胞类型所占比例矩阵H和细胞类型特异性的甲基化位点,当参考数据已知时它可以为估计得到的匿名细胞类型分配类别标签,已验证Tsisal在细胞类型估计个数、细胞类型比例估计、细胞类型标签分配上都取得了更加准确的结果。
第三步,当细胞类型特异性的位点和细胞类型比例矩阵得到后,我们可以使用现成的机器学习算法SVM来构建疾病预测模型。SVM算法的求解可以转化为凸二次规划问题:
(1)
(2)
确定分离超平面
和分类决策函数
,其中x为训练向量,y训练向量的标签,
,
,
为凸二次规划问题最优解的一个分量。如何选取核函数是设计SVM分类器的关键步骤,根据DNA甲基化芯片数据特点,本文选择灵活性高、决策边界多样、参数少的高斯核函数:
(3)
一方面,我们可以使用细胞类型特异性位点所构成的甲基化谱矩阵
来预测疾病状态Z。另一方面,我们可以使用细胞类型比例矩阵H来预测疾病状态Z。由于没有独立的测试集,本文采用十折交叉验证来评价模型的性能,具体来说为:将肿瘤组织样本平均分成十份,每次选取9份作为训练集,1份做测试集,将十次预测结果的均值作为最终的结果,本文选取准确率(
)来度量模型的性能,高的准确率说明模型有更好的性能。
3. 肺腺癌(LUAD)与肾透明细胞癌(KIRC)样本的实证分析
本文以TCGA数据库中LUAD和KIRC的Illumina Infinium 450 K微阵列数据作为研究对象,其中LUAD有466个肿瘤样本和32个正常样本,KIRC有325个肿瘤样本和160个正常样本,参考面板数据为Reinius等 [11] 提供的六种细胞类型(CD8T、CD4T、CD56NK、B细胞、单核细胞(Mono)和粒细胞(Gran))的450 K甲基化数据。
首先,我们对LUAD和KIRC的甲基化数据进行预处理,删除在所有样本上取值全是缺失值的特征,并利用Combat [12] 去除样本之间的批次效应。由于RefFreeEWAS和TOAST算法需要预先进行特征选择,因此我们对于RefFreeEWAS算法选取变异系数(CV)最大的前1000个特征,对于TOAST选取方差最大的前1000个特征。
其次,我们利用Tsisal来估计LUAD和KIRC所包含细胞类型的个数,其估计值均为9。在细胞类型个数为9的假定下分别利用TOAST包 [13] 中的Tsisal函数、csDeconv函数,RefFreeEWAS包 [14] 中的RefFreeCellMix函数对以上两种癌症类型进行反卷积分解估计细胞类型组成比例矩阵。由于Tsisal在有参考数据的前提下可为匿名估计的细胞类型分别类别标签,因此我们在0.55的阈值下,为估计得到的匿名细胞类型分配了类别标签。图1(a)显示了LUAD利用Tsisal估计出的9种细胞类型的比例,其中Mono细胞在混合样本中比例最高,占比接近40%,CD8T占18%,Gran占7%,该结果与Lin [13] 的结果相一致。图1(b)显示了KIRC利用Tsisal估计出的9种细胞类型比例,可以看到KIRC主要包含CD8T、CD56NK这两种免疫细胞,其中 CD56NK占比接近30%,这些免疫细胞也被确定为肿瘤中相对更丰富的免疫细胞类型 [15]。

Figure 1. Boxplot of cell type proportions estimated by Tsisal of LUAD and KIRC
图1. LUAD和KIRC利用Tsisal估计的细胞类型组成比例的箱线图
由于肿瘤组织样本中真实的细胞类型组成比例是未知的,因此我们将Tsisal估计的细胞类型比例与TOAST和RefFreeEWAS估计的结果进行一致性分析来说明Tsisal估计的准确性。图2(a)显示了LUAD上Tsisal估计的Cancer cell 4所占比例和TOAST估计的Cancer cell 4所占比例的散点图,其相关系数高达0.8,9种细胞类型上两种方法估计的平均相关系数大于0.5。图2(b)显示了KIRC上Tsisal估计的Cancer cell 3所占比例和TOAST估计的Cancer cell 3所占比例的散点图,其相关系数高达0.7,9种细胞类型上两种方法估计的平均相关系数大于0.4。这些结果说明了Tsisal估计的细胞类型所占比例和TOAST的估计结果具有高度的一致性。进一步地,我们将所有免疫细胞所占比例求和作为正常细胞所占比例,将所有癌症细胞所占比例求和作为癌症细胞所占比例,由此计算出每一个肿瘤样本的纯度数据,将该结果与经典的肿瘤纯度估计算法InfiniumPurify进行一致性分析。图2(c),图2(d)分别显示了LUAD、KIRC上两种算法估计的肿瘤纯度散点图,它们的相关系数分别为0.75和0.64。此结果进一步说明了Tsisal估计的细胞类型比例的可靠性。
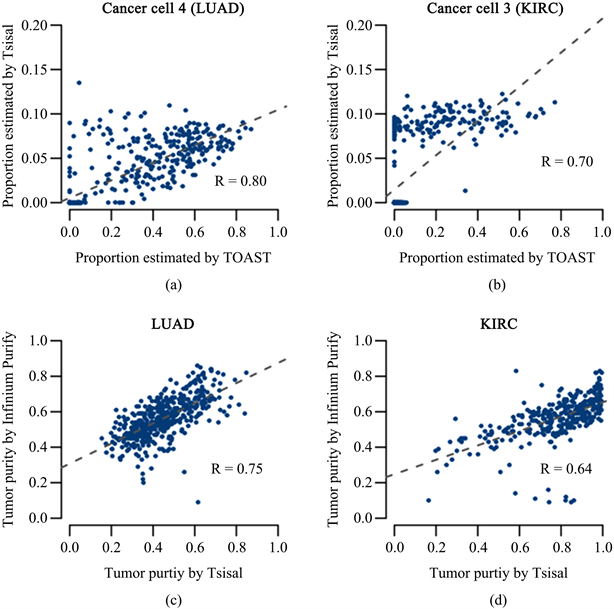
Figure 2. (a) Scatter plot of the estimated cancer cell 4 proportions between Tsisal and TOAST from LUAD; (b) Scatter plot of the estimated cancer cell 3 proportions between Tsisal and TOAST from KIRC; (c) Scatter plot of the estimated tumor purities between Tsisal and TOAST from LUAD; (d) Scatter plot of the estimated tumor purity between Tsisal and TOAST from LUAD
图2. (a) LUAD上Tsisal估计的Cancer cell 4所占比例和TOAST估计结果的散点图;(b) KIRC上Tsisal估计的Cancer cell 3所占比例和TOAST估计结果的散点图;(c) LUAD上InfiniumPurify估计的肿瘤纯度与Tsisal估计的肿瘤纯度的散点图;(d) KIRC上InfiniumPurify估计的肿瘤纯度与Tsisal估计的肿瘤纯度的散点图
由于Tsisal可输出每一个细胞类型特异性的CpG位点,因此对于每一个细胞类型我们选取特异性最大的前3个CpG位点来判断这些位点是否是细胞类型特异性的。图3(a)显示了LUAD中Cancer cell 1在cg00867626位点高甲基化,Cancer cell 3在cg16545496位点也出现高甲基化。图3(b)显示KIRC中Cancer cell 2在cg11474317位点表现出高甲基化,CD8T在cg13755570位点也表现出高甲基化。这些位点具有更为重要的生物学意义,也是我们进行疾病诊断预测的重要生物学标志物。
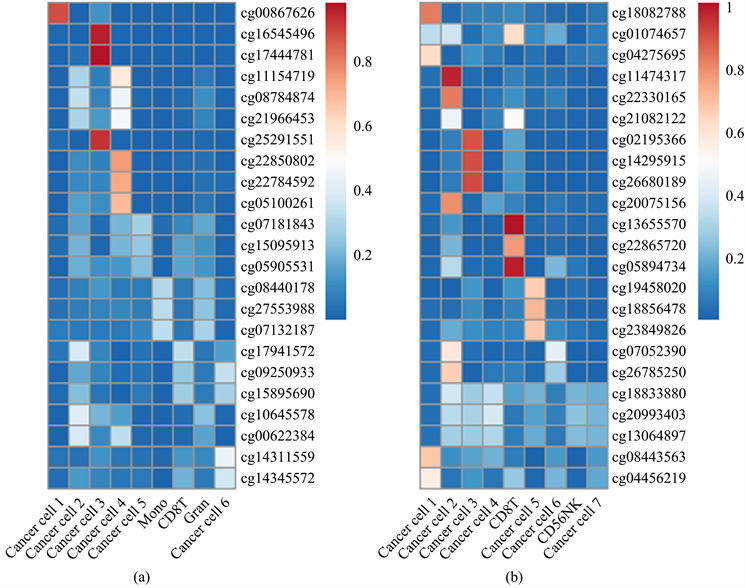
Figure 3. Heatmap of methylation profile data of cell type specific sites identified by Tisal on LUAD and KIRC
图3. LUAD和KIRC上Tisal识别出的细胞类型特异性位点甲基化谱数据的热图
以上分析结果证明了Tsisal所估计的细胞类型比例矩阵准确,其识别的位点也是细胞类型特异性的,因此最后我们将Tsisal估计得到的肿瘤组织中各细胞类型所占比例和细胞类型特异性的甲基化位点作为生物标志物,利用SVM算法进行疾病预测分析。具体做法为:将每一个细胞类型在样本上所占比例矩阵(或细胞类型特异性的甲基化位点在所有样本上的取值矩阵)作为自变量,将每一个样本的疾病状态作为因变量,利用R包el071中的svm函数构建模型,利用predict函数进行预测分析。图4(a)显示了LUAD上利用CV、TOAST和Tsisal选取细胞类型特异性的CpG位点(marker)作为SVM的输入来预测疾病状态时的准确率,CV和TOAST的准确率分别为0.92和0.96,而Tsisal的准确率高达0.99;当选择细胞类型估计比例(proportion)作为SVM的输入来预测疾病状态时,RefFreeEWAS的准确率为0.94,TOAST和Tsisal的准确率分别为0.98和0.99。尽管Tsisal与TOAST准确率较为接近,但Tsisal的运行时间是TOAST的六分之一。图4(b)癌症类型KIRC上取得了与图4(a)相类似的结果。这些结果表明利用反卷积算法Tsisal和SVM算法结合来构建疾病的预测模型具有较高的准确性、鲁棒性和运行效率。
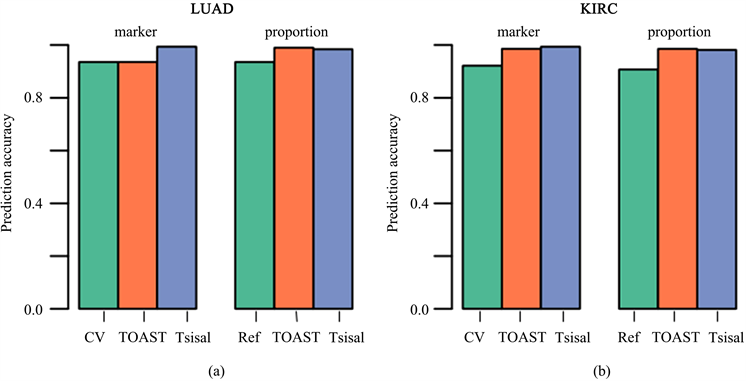
Figure 4. Accuracy of disease prediction under different biomarker selection methods on LUAD and KIRC
图4. LUAD和KIRC上不同生物标志物选取方法下疾病预测的准确率
4. 结论
本文针对临床肿瘤组织的甲基化数据构建了疾病的预测模型,该模型在正态分布假设下利用不依赖参考基矩阵的反卷积算法Tsisal对肿瘤组织进行分解,估计得到肿瘤组织中各细胞类型所占比例和细胞类型特异性的甲基化位点,将识别出的细胞类型组成比例和细胞类型特异性的甲基化位点作为生物标志物,利用机器学习中的SVM算法进行疾病预测,并利用十折交叉验证来评价算法的性能。TCGA数据库中肺腺癌、肾透明细胞癌甲基化数据分析表明,所提模型取得了优于TOAST和RefFreeEWAS的分析结果。
基金项目
研究得到了国家自然科学基金(61902061)、江西省自然科学基金(20212BAB202001)和江西省科技厅项目(GJJ200704)的支持。
NOTES
*通讯作者。