1. 引言
乳腺癌是当今临床治疗中一种常见的恶性肿瘤,其发病率逐年上升。世界卫生组织GLOBOCAN 2018年全球癌症统计报告显示,乳腺癌位居全球女性癌症发病率和死亡率的第1位,它严重威胁着女性的身心健康 [1]。女性乳腺癌已经成为一个全球性的重点公共医疗问题,为解决这个问题,药物研发领域对大量的候选药物进行了研究分析。研究发现,乳腺癌的发展与雌激素受体α亚型(Estrogen receptors alpha, Erα)息息相关,生物实验表明ERα在治疗乳腺癌中有极其重要的地位。截止到现在,医疗界的专家学者在ERα表达的病患中,通过控制雌激素受体活性进而控制患者体内的雌激素水平,起到治疗乳腺癌的作用。因此可以利用ERα的特性,研究针对性强的抗癌靶细胞药,与此同时,乳腺癌研发的备选药物也可以是拮抗ERα的活性化合物 [2]。
一个化合物想要成为治疗乳腺癌的候选药物,需要满足三点,良好的生物活性、良好的药代动力学性质以及安全性,记为ADMET (Absorption吸收、Distribution分布、Metabolism代谢、Excretion排泄、Toxicity毒性)性质,前四个名词代表着化合物在生物体内浓度与时间的变化关系,保证了药代动力学性质,T是观察化合物是否产生毒性物质,浓度是否可以接受,保证了药物的安全性 [3]。抗乳腺癌候选药物从研发到投入使用的过程中,如果仅仅采用实验的方式去评估化合物的生物活性、药代动力学性质和安全性,需要花费大量时间成本和经济成本。因此,在药物研发中,为了避免不必要的资源浪费,通常采用建立化合物活性预测模型的方法来筛选潜在活性化合物。研究机构通过把体外研究技术和计算机运算模型结合起来,针对与疾病相关的某个靶标(此处为ERα),收集一系列作用于该靶标的化合物及其生物活性数据,然后选取一系列分子结构描述符作为自变量,化合物的生物活性值作为因变量,构建化合物的定量结构–活性关系(quantitative structure-activity relationship)模型,然后使用该模型预测具有更好生物活性的新化合物分子,或者指导已有活性化合物的结构优化 [4]。此外,如果一个化合物的ADMET性质不佳,比如不容易被人体吸收,或者在体内代谢速度太快,或者具有某种毒性,那么其仍然难以成为药物,因而对其进行药物ADMET性质优化也是非常有必要的 [5]。
二十一世纪以来,随着信息技术的飞速发展,数据挖掘技术已经广泛应用于医药学研究,目前最常用的研究方式是利用计算机辅助的人工智能算法对药物生物活性和ADMET性质进行预测分析,研究学者通过数据拟合数学模型,进而通过优化的方法得到最优解,增加了研究的价值性,大大降低了研究成本,提高研发成功几率,且更有利于对候选药物在生物体内发挥的作用进行探索,有效避免因药物产生的副作用和毒性导致的人体疾病,可指导临床治疗时的合理用药 [6]。由此可见,使用计算机辅助的人工智能算法进行理论预测抗乳腺癌候选药物的生物活性和ADMET性质是极具现实意义的。
本文基于重构损失的图神经网络进行建模分析,凭借强大的端到端的学习能力,这类模型可以非常友好地支持有监督的学习方式。与较为常见的随机森林算法相比,使用图神经网络,用预先计算的分子描述符(或特征)来构建分子特性预测模型,其性能有明显提高,计算时间大幅度缩短。在运用XGBoost算法求解时,通过CART回归树模型进行拟合运算,激活函数得出目标函数值和对应的最优值。
2. 数据收集
针对乳腺癌治疗靶标ERα,从阿尔伯塔大学的DrugBank药物分子数据库中获取了1974种化合物对乳腺癌治疗靶标ERα的生物活性和ADMET性质数据,DrugBank数据库是一个整合了生物信息学和化学信息学资源,并提供详细的药物数据与药物靶标信息及其机制的全面分子信息,包括药物化学、药理学、药代动力学、ADME及其相互作用信息。相比于其他数据库来说,该数据库的信息更加准确,所有的药物靶点信息均是实验验证过的。
3. 筛选主要的分子描述符
3.1. 数据处理
针对收集到的1974个化合物对ERα的生物活性值的描述,从提供的729个分子描述符信息中选出 20个对生物活性影响最大的分子描述符。为方便建模计算,我们首先将所有变量数据进行编码,将变量按照从1~729的顺序编号,即表1所示:

Table 1. Sequential numbering feature parameters
表1. 顺序编号特征参数
在此编号的基础上,我们即有了一个1974 × 729的样本矩阵,并根据此矩阵建立模型并进行求解。由于传统的分类算法在低维度的数据集上面能够获得比较理想的分类效果,但是在高维度的数据集上它的分类性能则会出现较大的下降。高维数据结构复杂,包含更多的非信息和噪声,随机森林(Random Forests, RF)算法可以在对样本分类的同时给出变量的重要性评分(Variable Importance Measures, VIMs)作为特征筛选的依据 [7]。RF采用了特征子空间来构建模型,所以构建的模型难免会混杂很多的噪音,而利用这些包含噪音的模型进行预测分类将会降低随机森林算法的分类效果。使得VIMs不稳定,而那些真正对分类有意义的变量可能会因在筛选后得到的变量子集中排序较后而无法被选入 [8]。遗传算法(Genetic Algorithm, GA)是按照随机搜索策略进行特征筛选的,可以由不同的染色体提供多样化的特征筛选结果 [9],采用适当的遗传算法与随机森林模型相结合将有可能降低“噪声”对筛选结果的影响,同时保证筛选得到的特征变量集有较小的假发现率(False Discovery Rate, FDR)。所以本文考虑使用基于遗传算法的随机森林模型(GARF),用于1974个化合物的729个分子描述符的筛选,而最终挑选出前20个对生物活性最具有显著影响的分子描述符。
3.2. 基于遗传算法的随机森林模型筛选分子描述符
GARF采用RF模型对变量在样本分类中的作用进行评价,以Permutation方法确定特征筛选界值作为最终确定特征变量的依据。为减少噪声变量对RF变量评价结果的干扰,每个RF模型仅包含由GA算法选取的部分变量,并且在遗传过程中加入了变量筛选步骤以进一步降低噪声变量影响,尽量减少RF模型过拟合的可能。遗传过程内的变量筛选中,采用Permutation方法获得组间无差异变量重要性评分的经验分布,根据该经验分布自适应确定变量筛选界值 [10]。使用Python应用遗传算法–随机森林模型,运行程序得到729个变量的贡献度排名,由大到小将贡献度排序。考虑到下一步的高相关性滤波操作会对进一步变量进行降维,故先筛选出排名处于前26的变量,如表2所示。
Table 2. Filtered 26 feature parameters
表2. 筛选后的26个特征参数
为了提高遗传算法的收敛性,采用最优保留策略,将最大适应度的个体直接保留到下一代。每次更新种群时将群体中最差的个体进行替换成上一代的最优个体,以防止当前种群中适应度较好的个体被淘汰。将每一代中获得的变量评价结果取中位数,作为GARF特征筛选方法对变量的最终评价,记为
。计算最后一代种群中每条染色体中包含的基因个数;y取其平均值记为
,作为Permutation抽样参数;z从数据集中随机抽取
个变量,将分类标签随机打乱,建立随机森林模型,记录变量重要性评分,重复进行
次,共获得2000个变量重要性评分;以上述2000个变量重要性评的百分位数
或
作为GARF算法特征筛选界值,如
大于该界值则将该变量识别为特征变量。最后得出最终的20个参数指标,如表3所示;根据所提取的特征变量之间的距离相关系数计算结果对变量之间的相关程度进行可视化,如图1所示。可见选取的20个变量之间相关性低,独立性较好,这20个分子描述符能够尽可能地描述化合物的生物活性。

Figure 1. The strength of the correlation between the main variables
图1. 主要变量之间的相关性强弱
Table 3. Filtered 20 feature parameters
表3. 筛选后的20个特征参数
4. 基于图神经网络模型预测生物活性结果
在对分子描述符数据进行降维处理后,大大减少了数据量,鉴于化合物是由原子和化学键构成的,它们天然就是一种图数据的形式,图神经网络(GNN)能够自动化地同时学到图的特征信息与结构信息 [11],这一特点与分子描述符数据性质对候选药物影响方式的特点相吻合,因此可以选择图神经网络进行训练学习,并对50个化合物进行IC50_nM值和对应的pIC50值预测。本节分析基于图神经网络的生物活性值预测方法,并通过与相应的基于重构损失设计的损失函数结合起来实现无监督图表示学习,避免传统图神经网络有监督的学习方式。
基于重构损失构建的GNN模型进行生物活性值预测,其基本框架包括推断模型(编码器)、生成模型(解码器)、损失函数。
1) 推断模型
这里使用两个GNN对
、
进行拟合:
2) 生成模型
这里也简单使用了两个节点表示向量的内积来拟合邻接关系。
3) 损失函数
同样地,隐变量z的先验分布选用标准正态分布:
通过建立的化合物对ERα生物活性的定量预测模型,对50个化合物的生物活性值进行预测。得到的IC50值和对应的pIC50值,详见表4。
Table 4. IC50_nM value and corresponding pIC50 value prediction results
表4. IC50_nM值及对应pIC50值的最优预测结果
5. 基于XGBoost的分类预测模型
由于XGBoost的计算效率高、分类准确性强,且随着迭代次数的增加准确性大幅提升,对构建化合物的Caco-2、CYP3A4、hERG、HOB、MN的分类预测模型具有较好的预测效果,能有效降低预测误差,取得较高的预测精度 [12]。因此,本文将使用XGBoost算法构建化合物的ADMET预测模型,分别对5个指标进行预测分析。
通过CART回归树模型进行拟合计算,具体模型参考如下:
其中,n表示树数量;
表示函数空间F独立函数;
表示预测值;
表示输入i个数据;F表示CART总和。
迭代过程如下
XGBoost的目标函数如下所示:
目标函数近似为
激活函数:
其中,
为目标函数;
为一阶导数;
为二阶导数。
所得的最优
和目标函数值分别如下所示:
其中,
。
从图2的分子描述符变量间的相关性图可以看到,除了四个角是色块密集且颜色对应的相关系数绝对值接近1,相关度高,其余大部分区域色块较为稀疏,且颜色对应的相关系数值近于0,相关度低。所以综合看来,这些分子描述符之间的相关性较弱,以此为变量建模更具代表性与说服力。
从图3标准误差表中可得,黄线代表的是由常用的长记忆神经网络算法模型预测的结果,除了在横坐标为50和120处标准差偏低,大部分都是要比XGBoost模型预测的结果标准差大,由此可以看出本文所采用的XGBoost模型的预测效果要优于长记忆神经网络(LSTM),更准确更有说服力。
根据所构建的Caco-2、CYP3A4、hERG、HOB、MN的分类预测模型,可以由化合物分子的结构式对新化合物的ADMET性质进行相应预测,从而判断新化合物的性质好坏,对药物性质判断提供一定的参考价值。
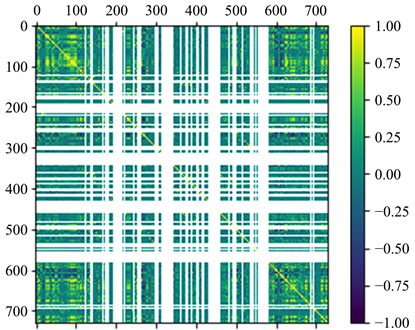
Figure 2. The strength of the correlation between the main variables
图2. 主要变量之间的相关性强弱

Figure 3. RSME percentage for LSTM and XGBoost
图3. LSTM和XGBoost的RSME百分比
6. 结论
针对抗乳腺癌候选药物研发过程中的生物活性和ADMET性质预测问题,本文提出的两个预测模型精度、鲁棒性表现优秀,采用的方法结合了遗传算法和随机森林,与一般的方法相比,预测的结果更加均衡,降低了单一方法预测结果产生的偏差。对于研制乳腺癌的治疗候选药物,减少乳腺癌的致死率以及防治其他肿瘤疾病等人体生命健康的研究具有一定的指导性。