1. 引言
在病理学、生物信息学和生物医学的研究中,基因和环境的交互效应对许多复杂疾病的预测、治疗和药物开发有着重要影响。全基因组协会(Genome-Wide Association Studies, GWAS)的研究已经识别出了许多基因效应(如基因的表达、甲基化、单核苷酸多态性),这些基因效应可以提供理解疾病和癌症的复杂机制的路径。也有研究表明环境和临床因素(如吸烟状况、空气污染、营养摄入等)同样与癌症有关。然而,对于许多复杂的疾病,单一的基因效应或单一的环境效应不能解释预测结果的全部变异。有许多癌症方面的例子可以体现基因–环境交互项的重要性。例如,携带COX-2基因单核苷酸多态性(基因效应)的人增加鲑鱼类鱼的摄入量(环境效应)会降低患前列腺癌的风险 [1]。
基因–环境的交互作用被定义为单个或多个基因因素和环境因素的协同效应,这种协同效应不能通过上述基因因素或环境因素的边际效应来解释 [2]。为了有效的识别出基因主效应及其与环境的交互效应,变量选择方法得到了广泛的发展。现有的交互项选择方法总体上分为两类。一种方法是,先采用边际分析选取所有的主效应,然后将对应的相关交互项加入到模型中。这些方法计算成本低,但效率也较低,因为它一次只分析少量的基因效应。另一类方法采用联合分析,通过层次约束、收缩方法、惩罚函数或不等式约束等方法,可以同时识别主效应和交互效应。这类方法不可避免地具有较高的计算成本 [3]。
尽管许多统计方法已被应用其中,但在识别基因–环境交互作用的研究中仍存在一些挑战。首先,基因数据的高维度和大样本容量导致计算成本较高。此外,由于有不同的方式和工具来测量评估来自不同来源的效应,数据具有异质性的特点。例如,某些空气污染的粒子大小和化学组成成分在不同地区是不同的 [4]。具有异质性的数据集在预测变量和响应变量中会出现异常值或者重尾性质等数据不平衡的特性 [5]。第三个挑战是数据污染和模型错误划分。通常模型会假设样本中没有异常值,但实际上数据中有异常值和杠杆点。而且大多数方法都会预设一个特定的参数或半参数模型。潜在的模型错误选择会导致估计偏差和分析结果不准确。这就要求模型具有稳健性,能够有效地处理数据污染和模型的误判。
Wu等人使用LASSO惩罚建立了加速失效时间(Accelerate Failure Time, AFT)模型,该模型是一种稳健的服从层次结构的变量选择方法,可以处理脏数据 [6]。Zhu等人提出了一种分阶段回归方法和渐进式惩罚项选择基因–基因和基因–环境的交互作用。这种方法计算速度快,同时遵从了强层次结构 [7]。Ma等人采用加权最小二乘方法和一组稀疏惩罚来识别交互效应,这种方法降低了计算成本且有较好的预测性 [8]。Ren等人提出了一种基于Spike-and-slab先验的半参数贝叶斯模型,可以同时探索线性和非线性的相互作用 [9]。Xu等人利用截尾分位数偏相关方法(CQPCorr)建立分位数回归模型,对长尾数据和不平衡数据进行G-E交互效应分析 [10]。
在本文中,我们建立了一种基于AFT模型并利用加权加权最小绝对偏差损失(Least Absolute Deviation, LAD)和平滑剪切绝对偏差(Smoothly Clipped Absolute Deviation, SCAD)惩罚函数进行基因–环境交互作用的变量选择方法。这种方法对于高维和删失的生存数据下的变量选择有很好的稳健性。本文采用的LAD损失函数可以适应数据中的污染和长尾误差,从而达到稳健性的目的。此外,SCAD惩罚函数具有无偏性、稀疏性和连续性的特点,对应的解收敛于局部最小值 [11]。三种误差分布下的数据仿真和乳腺癌数据案例研究表明,在基因–环境交互效应的识别效果方面,本文所提出方法的表现优于其他方法。
2. 基于SCAD惩罚函数的AFT模型
考虑一个包含n个独立同分布样本的数据集,其中有p个基因效应和q个环境/临床效应。对于第i个样本,设
为感兴趣的响应变量,即生存时间的对数,
为p个基因效应,
为q个环境效应。我们采用加速失效时间(AFT)模型来描述具有删失性的生存数据。与其他生存分析的模型相比,AFT模型形式简单、计算代价较低,适合处理高维数据。而且在AFT模型下,被估计的回归系数会有更清晰的解释 [8]。模型假设:
(1)
其中,
,
。值得注意的是,这里省略了截距,因为我们假定数据已经经过标准化,这使得响应变量的均值为零。
记
,
,
。对于第i个观测样本,符号
和
表示第j个基因效应对第i个观测的所有影响,包括主效应以及和环境/临床因素的交互效应。记
为截尾时间的对数。当生存时间为右删失时,令
为新的响应变量,
为标记当前样本是否删失的指标。
此外,我们还需要考虑主效应与交互效应的层次结构,以防一个交互作用被确定为相关变量而对应的主效应却不被识别为相关变量的情况出现。现有如下两种层次结构。强层次结构假设只有当对应的主效应都被选择时才能认为其交互作用可能相关。相比之下,弱层次结构假设条件更加宽松,只要确定了至少两种主效应中的其中一种,就可以认为对应的交互作用是相关的。Liu等认为在基因–环境交互作用检测研究中,倾向于使用强层次结构,因为环境因素往往起重要作用,且维数低、解释性强,不需要被筛选压缩 [8]。若考虑弱层次结构,需要考虑的交互项过多,模型复杂不利于解释。因此,我们采用强层次结构,用以下形式表示:
(2)
2.1. 构建目标函数
2.1.1. 损失函数
最小二乘法是一种不稳健的损失函数,因此本文考虑一个具有更强稳健性的L1损失函数。我们采用加权LAD函数的损失函数,既能达到鲁棒性的目的,又能适应脏数据。本文使用的权重是由Stute等人提出的Kaplan-Meier (K-M)权重 [12]。设
为响应变量
的分布函数的估计,则
可以被表示为
。其中,
被定义为:
(3)
然后,计算加权LAD损失函数如下:
(4)
2.1.2. 惩罚函数
Fan和Li等人提出了平滑剪切绝对偏差惩罚函数(Smoothly Clipped Absolute Deviation, SCAD),该惩罚在变量选择中具有良好的性质,例如:连续性、稀疏性、无偏性和高维情况下的渐近oracle性质。由于环境/临床因素的选择是基于广泛综合的先验知识,且数据的维度较低,所以不需要对环境/临床效应进行选择。因此,我们只对基因效应及其交互效应添加惩罚项。惩罚函数为:
(5)
其中,
是带有调节参数
的惩罚函数,可以通过以下函数计算:
(6)
正如Fan和Li所讨论的,SCAD惩罚项满足一个好的惩罚函数应有的三个理想特性:无偏性、稀疏性和连续性。也就是说,通过SCAD惩罚函数所得到的估计量将近似无偏,并自动同时将系数的极小值设为零,从而降低最终模型的复杂性,并具有连续性以保持预测的稳定性 [12]。此外,Kim等人还证明了在一定条件下SCAD惩罚函数的估计参数与oracle惩罚的解渐近等价,即估计参数具有渐近正态性 [13]。
2.1.3. 目标函数
我们将上述损失函数和惩罚函数代入如下稳健目标函数:
(7)
在这个目标函数中,
为与所有第j个基因效应相关变量的系数向量,包括作为第一分量的基因主效应和相应的其它基因–环境交互效应。而
只作为基因–环境交互效应的系数向量。对于第j个基因效应,我们采用群体水平和个体水平上的惩罚项来进行变量选择。在群体水平上,非零的
表示基因主效应或与环境的交互效应被识别到。而在个体水平上,非零的
只能说明了交互效应的存在。因此,基因主效应仅在群体水平上受到惩罚约束,而基因与环境的交互效应在群体和个体水平上都受到惩罚。只要一种交互效应被认为是相关的,其对应主效应就会进入到模型中。
2.2. 模型求解
由于目标函数是非凸的,因此采用迭代方法更新基因主效应系数和基因–环境交互系数直到收敛。首先,我们初始化主效应的系数,即
和
。然后采用CCCP (Convex-Concave procedure)算法求解
。其次,同样地,我们固定交互项系数
的值,并应用CCCP算法更新
和
的解。由于我们对惩罚函数施加了层次约束,使得主效应与交互效应之间存在强层次结构。然后重复这两个步骤,直到损失函数值足够小 [14]。
An和Tao提出了CCCP算法来解决非凸问题。该算法对初始值不敏感,且总是收敛于局部最小值,是一种鲁棒、稳定的算法。在高维情况下,SCAD可以同时实现模型选择的一致性和最优预测,而LASSO则无法做到这一点。具体算法如下:
SCAD罚函数可以被分解成凹函数和凸函数的和:
(8)
其中,在
是可微分的凹函数,
是凸函数。然后将目标函数改写为
(9)
我们注意到
的第二部分是一些凹函数的总和,而
第三部分是凸函数的总和。设初始化的解为 ,然后严格凸上界可以通过下面的式子计算:
(10)
函数
是一个分段二次函数,我们可以通过最小化
来更新
当前的值直至收敛。
3. 模拟分析
在基因效应的测定中,考虑到基因表达数据和SNP数据同时存在,我们可使用连续变量和分类变量来模拟。具体来说,基因表达数据是使用自回归相关结构从多变量正态分布中生成的。在该结构中,第i和第j个基因效应在强层次结构下具有相关系数
。在弱层次下具有相关系数
。SNP数据是分位数方法生成的3个水平的分类变量。我们根据第一个和第二个三分位数将数据分成三类。然后生成了具有3个水平的分类变量SNP数据。相似的模拟数据生成方法也被应用于其他的研究中,如Wu等人的研究 [6] 和Shi等人的研究 [15]。对于环境/临床效应的测定,我们也采用自回归相关结构下的多元正态分布来生成数据。共生成300个基因表达因子和SNP,环境因子5个,即主效应305个,交互效应1500个。生存时间的对数 由以下数据生成模型计算:
(11)
假定有15个基因主效应和20个基因–环境交互项与响应变量(生存时间的对数Y)相关。式(11)中的系数由均匀分布Uniform (0.6, 14)生成。删失率假定为0.3。我们考虑如下三种不同的分布作为误差项的分布:第一种是标准正态分布
;第二种是标准正态分布与柯西分布的混合
;第三种是T分布
。
除了记为LAD-SCAD的我们所提出的方法外,还增加了5种备选的方法来作比较,包括LAD-LASSO、LAD-Group、LS-SCAD、LS-LASSO和LS-Group。在LAD-LASSO和LAD-Group中分别采用LASSO惩罚项和Group LASSO惩罚项代替SCAD惩罚项。二者的区别在于LASSO会在群体水平和个体水平上都产生稀疏性,而Group LASSO在组内不存在稀疏性、只在群体水平上产生稀疏性。对于LS-SCAD,保留SCAD作为惩罚函数,用LS (即最小二乘法)代替LAD作为损失函数。LS是一个对异常值敏感的非鲁棒损失函数。以下是数据仿真的结果:

Table 1. Comparison of simulation results of six methods
表1. 六种方法的模拟结果比较
仿真结果如表1所示。表中所有的值都是100次重复模拟的平均值,括号中是标准差的值。TP/FP代表真/假阳性的总数。TP1/FP1表示交互作用的真/假阳性的数量。由于本文的目的是检测G-E的相互作用效应,因此TP和TP1是模型选择的关键标准。其他模型性能评估的方法有PMSE和RSSE。PMSE表示预测均方误差。RSSE代表平方误差总和的平方根,可以通过公式
来计算,其中
和
分布代表了系数
的真实值和预测值。所选变量的总数也显示在表中,记为N_VAR。
在三种误差分布下,比较六个不同模型的PMSE的值,我们可以发现带有最小二乘损失的模型,如:LS-SCAD、LS-LASSO和LS-Group,具有较大的PMSE值(大约为2)和方差。其他三个带有LAD的模型的PMSE值(大约为1)和方差较小。可以看出最小绝对偏差的方法(LAD)更加预测准确和稳健。
另外,我们注意到LS-LASSO和LS-Group是两个特殊的模型,其TP和FP值明显高于其他四个模型。虽然这两个模型可以识别出很多相关的效应,但模型中也包含了一些冗余效应,降低了模型的可解释性和简洁性。从表的第一部分我们可以看到,在误差服从标准正态分布的情况下,这两个模型将超过120个主效应和交互效应识别为重要的。因而有必要找到一些更有效的方法来压缩备选变量集中的变量。下面将分析其他四种方法。
在误差分布为标准正态分布
下,即数据未被污染的情况下,LAD-SCAD和LAD-Group表现更好,TP和TP1较高。但在两个模型中,LAD-SCAD方法的FP和FP1值较小,它选择出的不必要的变量较少。我们再比较LAD-LASSO和LAD-Group的FP和FP1的值,LAD-Group的值远大于LAD-LASSO的,这是因为LASSO会在群体水平和个体水平上都产生稀疏性,而Group LASSO只在群体水平上产生稀疏性,但在组内不存在稀疏性。Group LASSO惩罚函数的压缩性较差。
在误差分布为标准正态分布和柯西分布的混合分布下,数据存在污染,LAD-SCAD、LAD-LASSO和LAD-Group三种方法的TP和TP1值很相近。但LAD-SCAD的PMSE值(0.86)小于其他几种方法的PMSE值(1.12, 0.92, 2.05),有较好的预测性。
在误差分布为T分布t (2)下,LAD-SCAD和LAD-Group的TP和TP1的值较其他两种误差分布下更高。可以看出,特别是在数据存在重尾分布的样本不平衡情况下,LAD-SCAD和LAD-Group表现出了在基因–环境交互效应选择方面的优越性。但同时FP和FP1的值也高于其他两个模型(LAD-LASSO和LS-SCAD)。但为了更准确地识别交互作用,这仍然是值得的。我们还注意到,在三种误差分布下,LAD-SCAD的FP和FP1值都小于LAD-Group的,这说明当两种方法都选择了相似数量的正确相关协变量时,LAD-SCAD方法比LAD-Group方法更好。
此外,为了更直观地比较不同模型的变量选择性能,我们实现了数据可视化。
从图1可以看出,LAD-SCAD是一种合适的选择变量的方法,这种方法选择了尽可能多的基因–环境交互效应,有效压缩了变量,预测误差较小。LS-LASSO和LS-Group许多冗余的效应识别为相关的,这降低了模型的可解释性和简单性。LAD-Group也是一种有效的方法。但是,真阳性数值相近的情况下,但LAD-SCAD的假阳性数值比LAD-Group少。当两种方法都选择了相似数量的正确相关协变量时,LAD-SCAD模型选择的冗余变量更少。所以LAD-SCAD方法比LAD-Group方法更好。
4. 实证研究
本文利用来自北卡罗莱纳大学基因鉴定中心的乳腺癌(Breast Cancer, BRCA)基因表达序列数据集进行实证分析。该数据集共包含17815个基因效应和597个观测样本。为了便于比较,数据已经过标准化处理。我们根据突变概率从高到低对备选基因效应进行排序。由于突变较高的基因与癌症有较高的关联概率,因此选择前300个易突变的基因效应进行后续分析。环境/临床效应数据从癌症基因组图谱联盟下载,该数据由美国国家癌症研究所和美国国家人类基因组研究所共同提供。我们考虑了5种环境效应:确诊年龄、性别、突变计数、基因组改变比例和放射治疗。即我们有300个基因效应,5个环境效应和1500个交互效应。
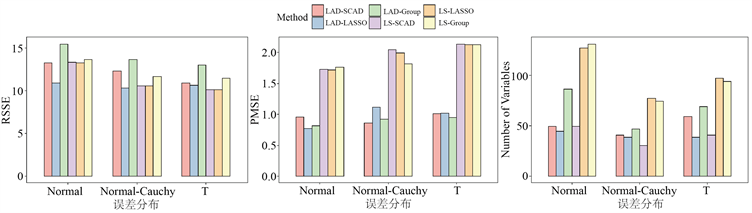
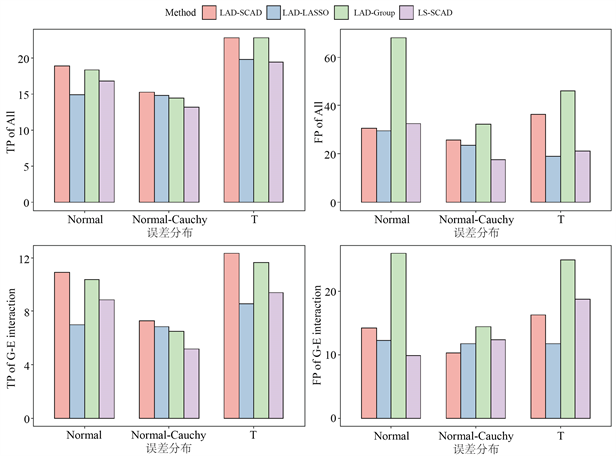
Figure 1. Histogram of simulation results of six methods
图1. 六种方法的仿真结果直方图
在此,本文采用模拟研究中四种更有效的方法来识别与乳腺癌相关的基因主效应和基因–环境交互效应,并比较其分析结果。LS-LASSO和LS-Group没有参与案例研究,因为这两种方法选择了很多冗余的变量,降低了模型的可解释性。
LAD-SCAD方法的变量选择结果如下:
如表2所示,我们所提出的LAD-SCAD方法共检测到35种效应,包括12种基因主效应和18种基因–环境交互效应。环境效应的估计系数分别为−0.095 (确诊年龄)、−0.119 (基因组改变比例)、−0.235 (突变计数)、0.191 (放射治疗)和0.047 (性别)。根据估计值的正负性质,这些结果是合理的。例如,较年轻的病人和基因组改变和突变较少的病人往往有较长的生存期。放射治疗对延长生存期有积极作用。与基因产生了最多交互项的环境变量是放射治疗,即放射治疗与多个基因效应共同作用对乳腺癌产生了影响。
Table 2. Results of variable selection analysis of BRCA data
表2. 乳腺癌数据的变量选择分析结果
其中有些基因效应已被证实与乳腺癌是相关的。例如,COL11A1的基因表达与乳腺癌的发生和进展过程相关,它的mRNA水平用于乳腺癌诊断预后的灵敏度高达81.31% [16]。USP9X可以影响乳腺癌细胞的生长和侵袭调节中心体的复制,为寻找潜在的乳腺癌治疗干预的靶点提供参考 [17]。UTRN基因表达被抑制后,细胞生长增殖的速度加快,所以UTRN对乳腺癌的干预治疗可以提供切入点 [18]。MYH9基因编码一种非肌细胞肌球蛋白II,影响乳腺癌细胞的生长、迁移、黏附、侵袭能力 [19]。
除了LAD-SCAD方法,我们还使用其他三个模型分析了BRCA数据。如表3所示,LAD-LASSO、LAD-Group和LS-SCAD识别出的变量数分别为27 (包括13个交互作用)、30 (包括14个交互作用)和38 (包括24个交互作用)。
Table 3. Result of BRCA data by four models
表3. 四种模型对乳腺癌数据的分析结果
从表3中可以看出,我们所提出的LAD-SCAD的预测误差是最小的,较为准确。LAD-LASSO和LAD-Group两种方法的预测误差较为相近,但由于LAD-LASSO比LAD-Group多在个体水平上产生了稀疏性,LAD-LASSO所选变量较少,模型更简洁。而LAD-Group选出了过多的与响应变量无关的冗余变量,降低了模型的可解释性。LS-SCAD的预测变量最大,准确性不如其他三个模型高。
5. 结论
在本篇文章中,我们提出了一个满足“主效应–交互项”强层次结构的交互作用识别方法。与现有的交互项选择方法相比较,我们根据生存数据不平衡的删失特性采用了加权LAD损失函数,并对群体水平和个体水平都添加了SCAD惩罚函数使得选出的效应服从强层次结构。然后采用CCCP算法对目标函数进行优化,从而得到主效应和交互项的系数估计。我们进行了数据模拟,在三种不同的误差分布下比较了该模型与其他五个模型的多个指标,从结果可以看出,与其他模型相比,我们提出的方法预测更准确、得到的模型更简洁。我们还收集了乳腺癌的基因表达数据(BRCA),利用所提出方法选出与乳腺癌相关的基因效应与其对应交互效应。仿真研究和实例研究都说明了该方法能准确、稳健地选择出合适的基因效应和基因–环境交互效应,且我们的方法能有效压缩备选的变量,选出的模型简洁、有较好的解释性。
基金项目
国家自然科学基金项目“高维数据变量间非线性交互作用的研究”(11571009);山西省应用基础研究计划“复杂环境下肿瘤免疫系统的动力学建模及致病基因识别”(201901D111086)。