1. 引言
能源是现代社会发展的基础,但能源的日益减少和环境的不断污染,已经严重影响到人类的生存和发展。为此开发和发展新能源已成为世界各国的主要目标。从目前新能源发展情况来看,氢能源无疑很有优势,首先氢气来源广泛,可以水解产生,轻质、燃烧热量高且不污染环境,这些优势使得人们一直非常关注氢能源的应用。尽管氢能源有很多传统能源不具备的优势,但是安全高效的储氢方式,是制约氢能源能否大规模利用的一个重大难题,为此全世界的科研人员进行了大量的研究。然而,到目前为止氢的安全储运技术还远远达不到实际应用的要求。团簇作为材料科学的一个重要分支,团簇储氢已经被广泛研究 [1] [2] [3]。而轻质铝团簇应用于储氢也是寻求合适储氢材料的一个研究方向 [4] [5] [6]。
在早先,Upton [7] 等人就已经研究了Aln (n = 2~6)与H2分子之间的相互作用。一些研究显示在纯的铝团簇中加入碳、硅、氮等非金属原子,团簇的结构以及一些物理化学性质会发生显著的变化。C. Ashman和Zhao [8] [9] 的研究都表明碳原子掺杂后的Al7C−团簇具有幻数结构,稳定性有了很大的提高,可以用来进行团簇的组装和凝聚。Ma [10] 等人用DFT理论研究了氢分子在Al4Sim (m = 2、3、4)上的吸附和解离,结果表明吸附能非常弱,而氢分子解离能垒达到0.76 eV。目前的研究表明过渡金属和碱金属修饰纳米团簇能获得较好的储氢能力 [11] [12] [13] [14],然而过渡金属的内聚能达到4.0 eV,很容易聚集在宿主团簇上,从而导致储氢效率下降,而碱金属的内聚能很小,可有效地避免这一问题。祁鹏堂 [15] 研究了碱金属Li原子修饰的C24团簇对氢分子的吸附作用,表明一个Li原子最多吸附3个氢分子,平均吸附能在−0.08~−0.13 eV之间。我们 [16] 也对Al7C团簇做了细致研究,并探讨了其与氢分子的反应活性,发现氢分子在Al7C体系上的吸附非常弱。因此我们对Al7C体系掺杂碱金属进行研究,结果H2在Li掺杂的Al7C+上的吸附能可以达到−0.151 eV。目前铝团簇储氢的研究主要集中在利用非金属元素掺杂,而对于碱金属元素修饰Al-C体系的研究很少,特别是利用Na修饰Al-C体系的储氢尚无报道。因此我们利用密度泛函理论 [17] 的B3LPY∕6-31g**方法计算分析了Al7C以及Na修饰的Al7CNa团簇的结构,最后优化得到Al7CNa-H2体系的稳定吸附结构。
2. 计算方法
Al7C和Al7CNa团簇的初始结构,参考了文献 [8] [9] [18] 给出的结构。同时我们也对所有可能的初始结构进行几何结构优化,在结构优化过程中没有对称性限制。我们采用密度泛函理论的B3LPY方法,选用的基组是加入极化函数ζ的6-31g**,计算得到的最稳定结构没有出现虚频,并且与文献给出的结果基本一致。在计算Al7CNa-H2体系的稳定结构时,方法和基组保持不变,H2分子在Al7CNa团簇上以不同吸附方式进行非限制性优化,计算了团簇与H2分子之间的吸附能。所有的计算是在Gaussian 03程序 [19] 下完成。
3. 结果与讨论
3.1. Al7C的结构
对于Al7C团簇的初始结构,我们引用了文献中的结构 [8] [9],图1给出三种Al7C团簇的稳定结构。三个结构的几何构型比较类似,都是六个Al原子构成一个三棱柱,剩余的一个Al原子处在三棱柱的一个侧面,C原子位于三棱柱内部。三个结构的键长和键角在表1中给出。表2列出了三个结构的结合能,可见阳离子团簇的结合能最小,由于阴离子团簇的几何结构发生较大的扭曲,所以结合能最大。
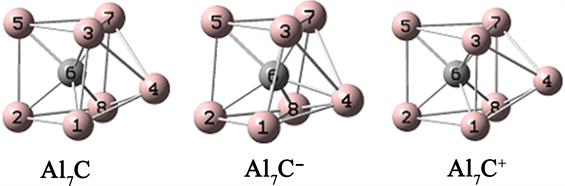
Figure 1. The stable structures of Al7C
图1. Al7C的稳定结构

Table 1. The structural parameters of Al7C
表1. Al7C团簇的结构参数
Table 2. Binding energy of Al7C and Al7CNa clusters (in eV)
表2. Al7C和Al7CNa团簇的结合能(单位:eV)
3.2. 碱金属修饰Al7C团簇的结构
根据文献给出的Al7CX (X = Li-Cs)的结构 [18],再依据前期计算结果,对Al7CNa团簇的结构进行了优化,结构如图2所示。从图2可以看出,Na原子引入后结合在三棱柱的下底面,三棱柱侧面的Al原子有较小的下移,结构的其他形变不是很明显。
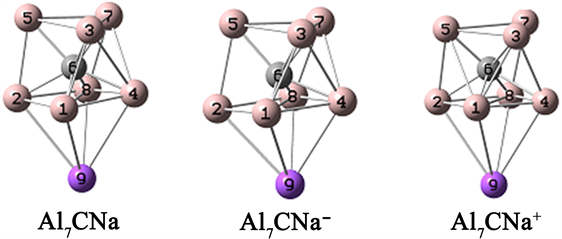
Figure 2. Stable structure of Na decorated Al7C clusters
图2. Na修饰Al7C团簇的稳定结构
结合能是判断团簇稳定性的一个很重要的指标,我们定义Al7CNa团簇的结合能为Al7CNa团簇的能量与Al7C团簇和Na原子的能量之差:
从表2可看出Al7CNa中性与阳离子的结合能比较接近均大于2.0 eV,而阴离子团簇的结合能仅为0.48 eV,明显小于中性以及阳离子的。从结合能的角度可以判定阳离子的稳定性最高,阴离子的稳定性最差。对比Al7C和Al7CNa,有一个很有意思的现象是它们的稳定性恰好相反。
3.3. 碱金属Na修饰Al7C团簇的电子结构
通过分析团簇的自然键轨道(NBO),得到碱金属修饰后Al7CNa上电子的分布,表3是Al7C和Al7CNa中性、阴离子、阳离子团簇的能隙与NBO电荷。对于Al7C团簇,Al的3p电子往C原子转移,且三种结构转移的总电子数目相同,Al7C中性、阴离子和阳离子团簇中C原子上的电荷分别为2.72、2.68、2.76个电子。引入碱金属后的Al7CNa团簇中,Al的3p电子、Na的3s电子向C原子转移,鉴于碱金属最外层的电子向C原子转移,7个Al原子的3p轨道上失去的电子减少。通过表3可见,修饰前后C原子上总电荷大致相同,均在2.70个电子左右。Al7C中阳离子的能隙最大为2.65 eV,中性、阴离子的分别为1.34 eV、1.83 eV。相反,Na修饰以后中性团簇的能隙最大为2.60 eV,阴离子与阳离子的能隙分别为1.32 eV和1.16 eV。
Table 3. Energy gap and natural bond orbital (NBO) charges of Al7C and Al7CNa clusters
表3. Al7C及Al7CNa的能隙和自然键轨道(NBO)电荷
3.4. H2在Na修饰Al7C团簇上的物理吸附
关于Al7、Al7C团簇与小分子(如H2、D2、O2)的相互作用已有许多研究,我们前期的计算也表明Al7和Al7C对H2分子吸附非常弱,而且H2分子很容易被Al7团簇解离。当碱金属修饰后铝团簇的能隙发生了很大的变化,由于碱金属失电子能力较强,Al原子失去的电子明显减少且电荷的分布有较大变化,因此小分子与修饰后团簇之间的相互作用发生显著改变。
研究H2分子与Al7CNa团簇相互作用时,吸附体的初始结构为H2分子按一定取向放置在Na原子周围。对不同初始结构使用B3LPY∕6-31g**进行全优化。H2分子的平均吸附能为:
这里的E (Al7CNa + nH2)是Al7CNa体系和H2分子的总能量,E (Al7CNa)是Al7CNa体系的总能量,E(H2)是一个H2分子的能量,n是H2分子数目。
图3是H2分子在Al7CNa团簇上的吸附结构,H2分子最多吸附4个,以侧位方式吸附在Na原子周围。Al7CNa中性团簇吸附一个和四个H2分子吸附能分别为−0.046 eV、−0.025 eV。阴离子的吸附能最弱,仅为−0.008 eV和−0.014 eV。阳离子的吸附能最高,分别达到了−0.088 eV和−0.055 eV。Na原子与H2分子之间的距离及H-H键长能直观的反应出吸附能的强弱,吸附能越大,H2分子与宿主团簇之间的距离越小、H-H键越长。
在研究Na原子修饰Al7C团簇对H2分子的吸附机理时,根据NBO电荷分析,Na原子往C原子转移电荷,在H2分子接近带正电的Na原子时,Na原子产生的静电场使得H2分子发生极化,在极化作用下H2分子会吸附于Na原子周围,并且吸附能较小,因此团簇对H2分子的吸附属于弱的物理吸附。
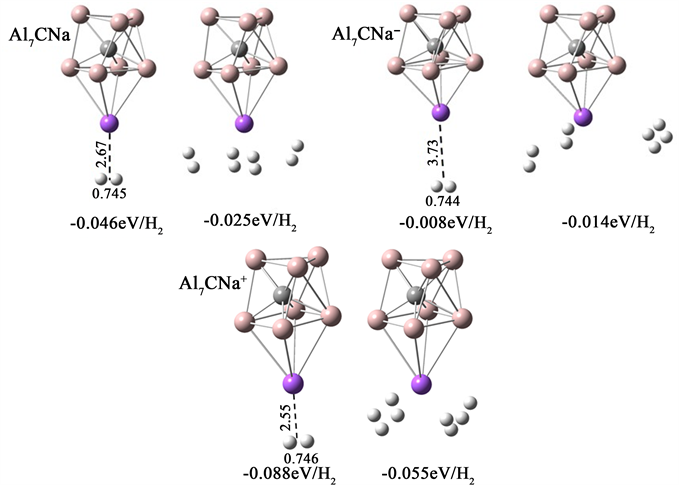
Figure 3. The stable adsorption structures of H2 on Al7C
图3. H2分子在Al7CNa团簇上的稳定吸附结构
4. 总结
本文应用密度泛函理论在B3LPY∕6-31g**水平上计算了Al7C、Na原子修饰的Al7C中性、阴离子、阳离子团簇的几何结构和电子结构。引入Na原子修饰后,对氢分子的吸附明显增强。Al7CNa阳离子团簇对氢分子的吸附能为−0.088 eV,最多可吸附四个H2分子,平均吸附能为−0.055 eV。Al7CNa团簇对氢分子的吸附都为弱的物理吸附。
基金项目
国家自然科学基金(11804264),新疆维吾尔自治区自然科学基金(2021D01B47),新疆理工学院校级科研项目(XLY1907)。
NOTES
*通讯作者。