1. 引言
DNA结合蛋白在细胞内外各种生命活动中扮演着重要角色,例如:转录调控、DNA复制、DNA包装、DNA修复与重组。这些独立折叠结构域中的蛋白质至少有一个结构基序,并且与DNA有亲和力 [1]。DNA结合蛋白或配体可以作为抗生素、药物、类固醇等多种生物化学物质应用于DNA的生物物理、生化和生物学研究中 [2]。1986年,Swiss-Prot数据库只包含3939条蛋白质序列 [3],但是截止2020年10月7日UniProtKB/Swiss-Prot已经收录了563,972条蛋白质序列。基于实验的DNA结合蛋白检测方法虽然有很多 [4] [5] [6] [7],但存在价格昂贵、检测时间长、设备要求较高的问题 [8]。因此,开发一种可以有效区分DNA结合蛋白和非DNA结合蛋白的计算方法具有十分重要的意义。到目前为止,有很多基于蛋白质结构和序列的计算方法来预测DNA结合蛋白。由于获取的限制,利用蛋白质结构预测DNA结合蛋白虽然准确但无法进行高通量注释 [9] [10]。
近年来,研究人员开发出很多基于蛋白质序列预测DNA结合蛋白的计算方法,例如:iDNA-Prot [11],DNA-Prot [12],DNA-binder [13],iDNA-Prot|dis [14],PSFM-DBT [15],DeepDRBP-2L [16]。为了从蛋白质序列中提取更多的生物信息,研究人员使用了20种氨基酸组成频率、进化保守信息、蛋白质二级结构和氨基酸物理化学性质等特征表示方法构造特征。同时,采用特征选择算法降低特征冗余和相关性,提高预测模型性能。此外,常用于预测DNA结合蛋白的机器学习分类算法包括随机森林(Random Forest) [11] [12] [17] [18]、支持向量机(support vector machine, SVM) [19] - [25]、Logistic回归(Logistic Regression) [26]、朴素贝叶斯(Naive Bayes) [17]、人工神经网络(Artificial Neural Network, ANN) [27]。支持向量机具有较高的稳定性和精确性,被广泛应用于DNA结合蛋白预测识别领域。
然而,如何从蛋白质序列中提取序列顺序信息或关键模式是最重要和最困难的问题。Huang等人 [19] 从图形表示和氨基酸物理化学性质角度提取信息并使用mRMR算法建立蛋白质功能预测模型;Zou等人 [20] 根据组成信息、氨基酸理化性质、进化保守信息和结构功能信息预测DNA结合蛋白,从全局序列描述、非局部序列描述、局部序列描述三个角度构建DNA结合蛋白特征并建立基于支持向量机的预测模型;Kumar等人 [22] 使用氨基酸组成和伪氨基酸组成作为输入特征建立β-内酰胺酶蛋白质预测模型,取得了较好效果;Liu等人 [28] 使用周氏伪氨基酸组成特征和基于距离变化的物理化学性质特征建立microRNA预测模型,并可以应用于计算生物学的许多领域。
蛋白质的结构和功能是由20种天然氨基酸的物理化学和生物化学性质定义的,这些氨基酸是蛋白质的组成成分。AAindex数据 [29] 库包含544组氨基酸物理化学性质指数,每组指数由20种天然氨基酸数值构成。因此,从AAindex数据库中找出可以有效区分DNA结合蛋白与非DNA结合蛋白的氨基酸物理化学性质具有重要意义 [30]。由于测量方法与设备限制,有13组氨基酸物理化学指数存在缺失值,无法进行分析。近年来,通过对AAindex数据库进行数据挖掘研究,研究者们提出了很多关于蛋白质功能的预测方法 [31]。
本文通过柱状图表示法从531个氨基酸物理化学性质中提取了531个特征,同时还考虑氨基酸组成对蛋白质功能的影响。但是551个特征向量中存在冗余和多重相关性,这会增加预测器学习的难度和降低模型的精确度。为此,本文采用了三种特征选择方法来减少551个特征向量的冗余信息,即基于LASSO的方法 [32]、基于过滤器的方法、基于包装器的方法,并结合支持向量机对DNA结合蛋白进行预测(5折交叉验证)。并且所选取的特征通过了t检验,证明对于识别DNA结合蛋白和非DNA结合蛋白具有统计学意义。
为说明grDNA-Prot方法的有效性,本文在独立数据集DNAiset和DNArset上进行了验证分析,并与其他现有DNA结合蛋白预测方法DNAbinder、iDAN-Port、DNA-port进行对比。其中,DNArset数据集中非DNA结合蛋白数量远大于DNA结合蛋白,与生物界中DNA结合蛋白与非DNA结合蛋白的分布情况相符。结果表明,grDNA-Port方法优于现有基于序列的方法(DNAbinder、iDAN-Port、DNA-port)。本文grDNA-Port方法的分析框架如图1所示。
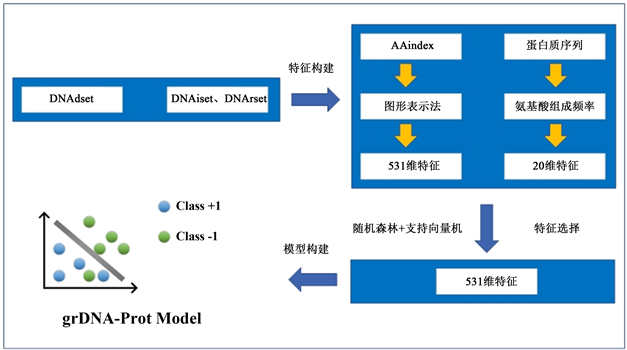
Figure 1. The research framework of this paper
图1. 本文研究框架
2. 材料与方法
2.1. 数据集
本文将DNAdset作为训练集,独立数据集DNAiset、DNArset作为验证集,这三个数据集被使用于多个DNA结合蛋白预测研究中 [20] [33]。训练集DNAdset包含231个DNA结合蛋白和231个非DNA结合蛋白,使用CD-HIT程序 [34] 检验该数据集内蛋白质序列相似性低于40%。Lin等人 [11] 在PDB (Protein Data Bank)数据库中检索关键词“DAN结合蛋白”得到2014年6月1日以后发布的97条DNA结合蛋白,并与199条非DNA结合蛋白混合形成独立数据集DNAiset。为模拟DNA结合蛋白在人体内分布情况,Kumar等人 [13] 在2007年发布了包含97个DNA结合蛋白和1500个非DNA结合蛋白的DNArset数据集。数据集DNAiset和DNArset中序列相似性小于30%。
2.2. 特征提取
特征包括氨基酸组成和氨基酸物理化学性质两类。许多氨基酸物理化学性质已经成功地应用于蛋白质长无序区、无序蛋白质结合残基的晶体结构注释、RNA结合残基的晶体结构注释、DNA结合残基的晶体结构注释等蛋白质结构和功能预测,例如氨基酸的疏水性、溶剂可及性、电荷和自由能。氨基酸的物理化学性质在蛋白质折叠和蛋白质与DNA相互作用中起着重要作用,本文使用531组氨基酸指数来表示氨基酸的各种物理化学性质。图形表示法 [35] 通过可视化方法提取蛋白质序列信息,是一种计算成本较低、无需对齐的方法,常常使用氨基酸的物理化学性质。本文在Yu等人 [36] 的工作基础上,用柱形表示法来表示蛋白质序列,将所建立的协方差矩阵特征值的最大值作为蛋白质序列的数值特征。因此,本文通过531组氨基酸指数得到531个数值特征来表示一条蛋白质序列。
下面将定义柱面图形表示法。20个氨基酸分布在圆柱体的底圆上,每个氨基酸在圆柱表面形成一条线。这种几何结构显示了氨基酸残基在蛋白质序列中的组成和分布。根据531个理化性质指标值对20个氨基酸进行排序。我们使用柱坐标来显示单位圆柱表面的蛋白质序列。
柱面坐标和笛卡尔坐标之间的转换如公式1所示:
,
,
(1)
其中,N表示蛋白质序列长度,
表示蛋白质序列中第n个氨基酸,
表示第n个氨基酸具体指标值。以AAindex数据库中氨基酸指数BHAR880101为例(若两个氨基酸指标值相同则按字母顺序排列),20个氨基酸的排序为:M < W < F < H < C < A < L < V < Y < T < I < N < K < Q < E < S < P < D < R < G。假设蛋白质序列为:
MKRRIRRERNKMAAAKSRNRRRELTDTLQAETDQLEDEKSALQTEIANLLKEKEKL
则蛋白质序列的柱状图表示如图2所示:

Figure 2. The cylindrical graphical representation of a protein sequence
图2. 蛋白质序列的柱状图表示
为了更直观地了解蛋白质序列中隐含的生物学特性,并分析它们的相似性与相异性,对蛋白质序列的圆柱形序列求协方差矩阵的最大特征值。协方差矩阵如公式2所示:
(2)
其中:
同时,定义
,
为协方差矩阵的特征值P。根据20个氨基酸的531个物理化学性质指标值,一条蛋白质序列通过上述方法可以转换成531个数值特征。另外,将蛋白质序列中20个氨基酸频率作为组成特征。
本文共得到551维特征用于DNA结合蛋白的预测,表示为
。并对特征进行标准化处理,标准化公式如式所示
(3)
其中,
和
分别为
的平均值与标准差。
2.3. 支持向量机(Support Vector Machine, SVM)
支持向量机是DNA结合蛋白预测领域中应用最广泛的机器学习算法。机器学习算法的原理是在高维特征空间中构造一个超平面,将数据点分为两类或者多类。基于径向基核函数的支持向量机算法已经广泛应用于蛋白质ATP结合位点预测等研究 [37]。径向基核函数如下所示:
(4)
其中
为径向基核函数的宽度。本文使用网格搜索和交叉验证确定参数C与
,两个参数的取值范围为
。
2.4. 性能评估
本文选择支持向量机算法作为分类器,使用五折交叉验证避免过拟合出现,并获得低均方误差的可靠结果。使用的评价指标为Accuracy (ACC)、Sensitivity, Precision, Specificity, F-measure and Matthews correlation coefficient (MCC) [38]。同时使用ROC曲线及ROC曲线下面积AUC评估分类器预测性能 [39]。这些指标由如下公式给出 [40]:
其中,TP和TN分别代表准确预测为DNA结合蛋白和非DNA结合蛋白的例数,FP代表预测为DNA结合蛋白的非DNA结合蛋白例数,FN代表预测为非DNA结合蛋白的DNA结合蛋白例数。TP、TN越高,FP、FN越低,代表模型预测效果更好。
2.5. 特征选择
为降低551维特征向量之间的冗余、提高预测模型精度,本文在训练数据集上(DNAdset)进行特征选择,得到一组非冗余的特征子集。本文比较了三种特征选择方法:LASSO方法、基于Filter的方法和基于Wrapper的方法,使用网格搜索和5折交叉验证确定每种方法中的最优参数,从而实现最优化预测性能(以AUC为评价指标)。第一种方法是利用LASSO回归方法得到特征间无线性相关性的特征子集。第二种方法先使用最大相关度最小冗余度(Maximum Relevance Minimum Redundancy, mRMR)方法对特征进行排序 [41],然后选择排名前200维特征。第三种方法是在基于Filter的方法基础上,根据随机森林(Random Forest, RF)算法得出特征重要性得分并对特征进行排序,然后通过支持向量机(Support Vector Machine, SVM)分类器得出的AUC值选择特征。具体方法为将重要性从大到小排序后特征依次放入当前模型中,若AUC值提高则特征被选进最优特征集,否则被排除最有特征集之外。
3. 实证分析
3.1. 特征选择结果
在这三种特征选择方法的基础上,采用基于AUC性能指标值的支持向量机分类器的5次交叉验证,得到了基于DNAdset的最优特征选择方法。详细结果见表1。

Table 1. The AUC of three feature selection methods based on 5-fold cross-validation for SVM on DNAdset
表1. 三种特征选择方法在数据集DNAdset上基于5折交叉验证和支持向量机的AUC结果
在表1中,结果表明,与其他两种基于支持向量机(SVM)分类器的5次交叉验证的特征选择方法相比,Wrapper-based (RF-SVM)特征选择方法具有更高的AUC值。在独立数据集(DNAiset和DNArset)上,采用支持向量机的最优参数和阈值0.49 (基于AUC值)来测试方法的性能。此外,利用Wrapper-based (RF-SVM)算法对选取的33个特征,在阈值0.44范围内得到ACC、MCC、精密度、灵敏度、特异度和F-测度值。具体评价结果见表2。此外,DNAdset、DNAiset和DNArset的ROC曲线如图3所示。
Table 2. Performance of prediction model on DNAdset, DNAiset and DNArset
表2. 预测模型在DNAdset、DNAiset和DNArset上的性能表现

Figure 3. The ROC curves of DNAdset, DNAiset and DNArset
图3. 在数据集DNAdset、DNAiset和DNArset上的ROC曲线
3.2. 两类特征预测性能分析
为了研究两类特征对DNA结合蛋白和非DNA结合蛋白的区分能力是否具有统计学意义,本文采用双侧t检验,显著性水平为0.05,33个特征中有24个(24/33 = 72.7%)具有显著性。因此,所选特征对区分DNA结合蛋白和非DNA结合蛋白具有统计学意义。
在选取的33个特征中,包含两种特征类型。其中属于氨基酸的物理化学性质的特征较多,为19个;属于氨基酸组成的较少,为14个。结果表明,融合后的33个特征会提高对DNA结合蛋白的预测性能(如图4所示),两类特征均对DNA结合蛋白的预测有重要影响。具体评价结果见表3。
在AAindex数据库(Tomii和Kanehisa,1996)中,理化性质指标基于最小生成树方法可以分为六类:Alpha and turn properties (A)、Beta propensity (B)、Composition (C)、Hydrophobicity (H)、Physicochemical properties (P)和Other properties (O)。将所选择的19个基于氨基酸物理化学性质的特征与六类属性对比表明(如表4所示),蛋白质序列中氨基酸残基的Hydrophobicity (H)、Physicochemical properties (P)和Alpha and turn properties (A),是区分DNA结合蛋白和非DNA结合蛋白的主要原因。
3.3. 与其他方法对比分析
为了验证grDNA-Prot方法的有效性,将grDNA-Prot方法与现有的三个web服务器(DNAbinder、iDNA-Prot和DNA-Prot)在两个独立测试数据集DNAiset和DNArset上进行了性能比较。详细结果见表5。对于DNAiset,grDNA-Prot对ACC、AUC、MCC和F-measure的性能评价高于DNAbinder和DNA Prot。

Figure 4. Comparison of the three feature types in DNAdset
图4. 在DNAdset中三种类型特征的比较
Table 3. The performance of the three feature types in DNAdset
表3. 在DNAdset中三种类型特征的性能
Table 4. The distribution of 19 features based on physicochemical properties of amino acids in six groups
表4. 19个基于氨基酸物理化学性质的特征在六类属性中分布情况
Table 5. Comparison of the predicted results by grDNA-Prot and other methods on DNAiset and DNArset
表5. grDNA-Prot在独立测试集DNAiset和DNArset上与其他方法的预测结果比较
此外,在DNArset的真实环境中,grDNA-Prot比DNAbinder、iDNA-Prot、DNA-Port获得更高的MCC,比DNAbinder、iDNA-Prot获得更高的F-measure。本文提出方法的性能接近iDNA-Port和DNA-Prot,但在两个数据集上的综合效果最优。这些结果表明grDNA-Prot方法可以有效地鉴定DNA结合蛋白。
4. 结论
DNA结合蛋白在细胞内外各种生命活动中起着重要的作用,现今已经研究出多种预测DNA结合蛋白的计算方法。本文提出的方法包含20维氨基酸组成频率特征和531维基于柱形图表示法的氨基酸物理化学性质特征,使用基于Wrapper的方法对融合后特征进行特征选择,选择出包含这两种类型的33维特征,最后建立了基于支持向量机的预测模型。同时发现,Hydrophobicity (H)、Physicochemical properties (P)和Alpha and turn properties (A)是区分DNA结合蛋白和非DNA结合蛋白的主要理化性质。因此,研究结果表明所选取的特征可以更好地解释绑定机制。
此外,通过在两个独立测试数据集(DNAiset和DNArset)上与其他方法(DNA-prot、iDNA-prot和DNAbinder)的比较,证明了grDNA-Prot方法的有效性。因此,grDNA-Prot可以相对准确地预测DNA结合蛋白。
基金项目
本文得到了河北省自然科学基金项目(F2019402078)、河北省高等学校科学技术研究项目(QN2018235)和河北省研究生创新资助项目(CXZZSS2021092)的支持,在此表示感谢。
NOTES
*通讯作者。