1. 引言
头孢菌素类抗生素中的高分子量杂质是引发速发型超敏反应的过敏原,高分子量杂质分为内源性杂质和外源性杂质,目前内源性高分子量的聚合物杂质是头孢类药物质量控制的重点。头孢菌素中高分子杂质类型主要分为只与母核有关的聚合反应和侧链参与反应的聚合反应两种类型。目前文献报道的聚合物主要有母核参与反应的二聚体、三聚体及侧链参与反应的二聚体和三聚体,还有少量的4~6聚体,其中有的抗生素聚合物杂质的分子量高达2000左右。聚合物杂质来自于生产过程,也可在产品出厂后的储运过程中形成,甚至也可因使用不当而产生。聚合物的形成在固体情况下与样品的水分有关,在溶液情况下与溶液的酸碱度密切相关,碱性条件更易形成聚合物。另外水分、时间、温度等对聚合物的形成有促进作用。
抗生素产品聚合物杂质的分离分析方法主要有反相高效液相色谱,凝胶色谱和离子交换色谱等。凝胶色谱一般选用Sephadex G-10作为β-内酰胺类抗生素高分子杂质的凝胶介质,经凝胶色谱柱分离后再用质谱分析聚合物杂质的结构信息。但是G10凝胶凝胶色谱结合质谱法分析聚合物杂质时,聚合物色谱峰的分离效果时常不令人满意,从而影响聚合物杂质的定量准确性。而合适的RP-HPLC方法对聚合物杂质的分离效果通常更好。
边磊等很早就用液质联用来检测头孢克肟中的有关物质 [1] [2] [3],赵晓东等人 [4] 较早报道了采用Sephadex G10色谱柱分离检测到头孢克肟中的一个聚合物杂质,石海英等 [5] 随后报道了用TSK-gel® G2500PWXL色谱柱分离检测到头孢克肟中的聚合物杂质,苏燕琼等 [6] 报道了用TSK gel G2000 SW XL色谱柱测定头孢克肟颗粒中的高分子杂质,证实头孢克肟双母核1、聚合物杂质B、头孢克肟二聚体F能较好的分离(双母核1对照品(广州牌牌生物科技有限公司,批号:20180518-01,含量为86.8%)、聚合物杂质B对照品(广州牌牌生物科技有限公司,批号:20181206-01,含量为90.34%)、头孢克肟二聚体F对照品(广州牌牌生物科技有限公司,批号:20190308-01,含量为88.46%))。黄艳等人 [7] 报道了用TSK-gel G2000 SW XL (7.8 mm × 300 mm,5 μm)色谱柱检测头孢克肟中聚合物的含量,在液相色谱中检测到三种聚合物杂质。李进等人 [8] 采用Phenomenex Gemini-C18型色谱柱,以0.5%甲酸水溶液0.5%甲酸乙腈溶液为流动相,进行梯度洗脱,建立RP-HPLC法分析头孢克肟聚合物,结果表明高效凝胶排阻色谱法分离头孢克肟聚合物杂质时,部分小分子杂质与聚合物共出峰,方法专属性差、定量准确性差;RP-HPLC法分析头孢克肟聚合物杂质时,能够检出2种头孢克肟二聚体、脱水二聚体等3种聚合物杂质峰。他们以头孢克肟降解溶液为供试品,采用柱切换LC-MS n法鉴定出两个四元内酰胺环开环后形成的二聚体(分子量906.08),一个头孢克肟二聚体,一个头孢克肟脱水二聚体,一个比头孢克肟二聚体的相对分子质量小109的头孢克肟二聚体降解物。
本公司一直致力于抗生素类杂质的检测、分离纯化和结构分析鉴定工作,对头孢克肟中的所有药典杂质均作了分析制备,还发现了更多的小分子杂质和聚合物杂质,其中我们发现了一个新的小分子杂质imp,并且发现它能形成聚合物杂质,并对其结构进行了推测。
2. 材料和方法
2.1. 仪器和试剂
Thermo Fisher Q EXACTIVE FOCUS ORBITRAP, UItiMate 3000Thermo Fisher Scientific。头孢克肟原料(批号:AP004-1906-2033,广州白云山制药厂)、乙腈为色谱纯(天津康科德),其他化学试剂(分析纯)从“西陇化工试剂有限公司”购买,色谱仪器用水为娃哈哈纯净水,分离制备用水为自制去离子水。
2.2. LC-HRMS分析检测头孢克肟中的imp杂质
色谱条件:色谱柱Thermo Fisher Hypurity C18,4.6 * 250 mm,5 μm;检测波长254 nm;流速1.0 mL/min;柱温30℃;进样量10 ul;流动相A:乙腈;B:0.1%甲酸水(洗脱条件:0 min (A:B = 10:90), 3 min (A:B = 10:90, 21 min (A:B = 40:60), 23 min (A:B = 40:60, 24 min (A:B = 10:90), 30 min (A:B = 10:90))。
质谱仪器参数:模式ESI+;喷雾电压:3.8 KV;金属毛细管温度:320℃;鞘气压力:30 arb;辅助气压力:10 arb;喷雾温度:120℃;S-lens RF level:55。
2.3. 分子排阻色谱法分析检测头孢克肟中的imp杂质
一维色谱条件:色谱柱TSK-gel G2000SWxl (300 mm × 7.8 mm, 5 um);检测波长254 nm;柱温20℃;流速0.6 ml/min;进样量20 μl;流动相A:pH 7.0磷酸盐缓冲液(1.02 g二水磷酸二氢钠和1.26 g十二水磷酸氢二钠,加入1000 ml超纯水,搅拌均匀,氢氧化钠调节pH 7.0):流动相B乙腈 = 95:5。采用Chromeleon 7.2工作站进行采集。等度洗脱(0 min (A:B = 95:5), 30 min (A:B = 95:5))
二维除盐色谱条件:色谱柱Welch(C18),Ultimate AQ-C18,5 μm, 3.0 × 50 mm;柱温:20℃;流速:0.300 ml/min;流动相:A:乙腈,B:0.1%甲酸水;洗脱条件(0 min (A:B = 1:99), 16 min (A:B = 1:99), 28 min (A:B = 100:0), 31 min (A:B = 100:0), 32 min (A:B = 1:99), 33 min (A:B = 1:99))。
十通阀切换时间:2D-LC-HRMS-MS进行头孢克肟原料药检测,先由下表确定聚合物组份1至组份4的保留时间,按表1进行2D-LC-HRMS-MS阀切换检测。

Table 1. Switching schedule of each impurity component
表1. 检测不同杂质成分时的切换时间
质谱参数:Thermo Fisher Q EXACTIVE FOCUS ORBITRAP,模式ESI+;喷雾电压3.8 KV;金属毛细管温度320℃;鞘气压力30 arb;辅助气压力10 arb;喷雾温度120℃;S-lens RF level55,勾选自动触发二级,工作站ThermoXcaliburQual Browser 4.0。
2.4. 溶液配制
精确称取原料药头孢克肟(批号:AP004-1906-2033) 10 mg溶于pH7.0的磷酸缓冲盐(精密称定磷酸二氢钾6.805 g和氢氧化钠0.940 g,加800 ml水溶解,先用饱和氢氧化钠调节pH至6.9,再用1.0 mol/L氢氧化钠调节pH值至7.0,搅拌均匀,用水稀释至1000 ml)超声溶解,用容量瓶定容至10 ml,使其浓度为约1 mg/ml,过滤后用于LC-HRMS检测。杂质对照品溶液:取imp杂质5 mg,用磷酸缓冲盐(pH 7.0)同上超声溶解,用容量瓶定容至100 ml,配成0.05 mg/ml溶液,过滤后用于LC-HRMS检测。杂质对照品+原料药溶液的配制:取imp杂质溶液适量滴加到头孢克肟原料药(批号:AP004-1906-2033)中,同上述方法配成0.05 mg/ml溶液。
2.5. 头孢克肟中的imp杂质的制备
1) 制备方法:90 g原料加入4.5 L水中,搅拌下滴加饱和磷酸氢二钾约300 mL,使之溶解完全,溶液总体积4.8 L,加入2.5 mL甲酸混匀,于C18制备柱中上样,用乙腈水溶液洗脱,经4次反复分离得到150 ml纯度97.53%的流份,冷冻干燥得到目标杂质。
2) 检测方法:杂质纯度检测方法用中国药典方法,柱温40℃,波长254 nm,流速1 ml/min,柱子InertSustain C18,5 μm,250 × 4.6 mm,流动相A:四丁基氢氧化铵溶液(取10%四丁基氢氧化铵溶液25 ml,加水1000 ml,摇匀,用1.5 mol/L磷酸溶液调节pH值至7.0),流动相B:乙腈。
3. 结果
3.1. LC-HRMS分析头孢克肟中的imp杂质
用LC-MS方法对imp杂质做了定位和分子量确认,测定结果为imp杂质在液相中的出峰时间是16.57 min,分子量为453.0 (图1,图2)。

Figure 1. Localization of imp impurities in cefixime by HPLC-MS
图1. 头孢克肟中imp杂质的HPLC-MS定位图

Figure 2. Localization of imp reference substance by HPLC-MS
图2. 头孢克肟imp杂质对照品的HPLC-MS定位
3.2. 从头孢克肟中分离纯化制备出的imp杂质确认
准分子离子峰454.04941 (M + H)和907.09071 (2M + H)其与头孢克肟imp杂质对照品保留时间及精密分子量基本一致,且头孢克肟imp杂质对照品滴加到头孢克肟原料中,头孢克肟imp杂质峰重叠且信号明显增加,在质谱检测中能观察到454.0491形成明显的双电荷(与第一个同位素相差0.5 Da),907.09071实为(2M + H)离子峰,确定为imp的二聚体。
另外原料药中未知杂质3与头孢克肟imp杂质相对保留时间基本一致,将头孢克肟imp杂质对照品分别添加到原料药中,杂质峰重叠,并且含量明显增高,确认头孢克肟imp杂质与未知杂质3为同一个物质 (图3~图5)。

Figure 3. HPLC analysis of impurities in cefixime
图3. 头孢克肟中的杂质的高效液相图

Figure 4. HPLC of imp impurity reference substance
图4. imp杂质对照品液相色谱图

Figure 5. HPLC of imp impurity with cefixime
图5. imp杂质加头孢克肟原料的液相色谱图
3.3. 分子排阻色谱法分析检测头孢克肟中的imp杂质
3.3.1. 一维色谱分析结果如表2所示
Table 2. One dimensional chromatographic analysis results
表2. 一维液相色谱分析结果
3.3.2. 二维色谱质谱检测结果(表3)
Table 3. 2D-HPLC-HRMS-MS results of component 3
表3. 组分3的二维液相色谱高分辨质谱结果
采用凝胶色谱法,通过2D-LC-HRMS-MS液质检测,结果表明,图6中组分3为头孢克肟imp杂质与聚合物(924)重叠,准分子离子峰分别为454.04871和925.10022;聚合物杂质的第三个峰中也能够提取出明显的质量数为907.09的峰,又证实了imp二聚体的存在。

Figure 6. Analysis of polymer impurities in cefixime by TSK gel permeation chromatography
图6. 头孢克肟中聚合物杂质TSK凝胶色谱分析
3.4. 杂质组份3(imp)的结构确认
组份3为头孢克肟imp杂质,LC-HRMS得到准分子离子峰(M+H)为454.04871,理论精密分子量为454.04911,实测值与理论计算结果相符,推断其结构如下(图7):
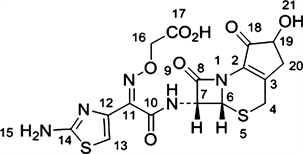
Figure 7. Molecular structure formula of impurity imp
图7. 杂质imp的分子结构式
组份3高分辨二级裂解碎片与推测结构一致,且头孢克肟imp杂质对照品的2D-LC-HRMS-MS质谱同位分析中保留时间、一级精密分子量及二级裂解碎片相同,故能够确认组份3为头孢克肟imp杂质,分子量为453.0。另外杂质imp的13CNMR,1HNMR数据也与该化合物的结构相符合。
3.5. 头孢克肟中imp的二聚体聚合物杂质结构推测
经Chemical office 软件计算,杂质imp二聚体聚合物的结构推测如下(图8):
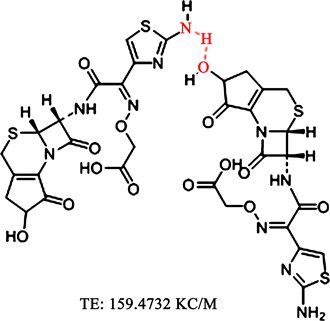
Figure 8. Inferred impurity structure of imp dimer based on intermolecular energy calculation
图8. 根据分子间能量计算推测的imp二聚体杂质结构
两个单体小分子(杂质imp)之间以分子内氢键的形式结合,这一结构在所有几个可能存在的结构中能量最低,相对稳定性最高。
4. 结论
采用LC-HRMS和2D-LC-HRMS-MS均能检测到头孢克肟中的新杂质imp,反相液相色谱法的分离度优于凝胶色谱层析法,凝胶色谱层析法在聚合物杂质分离确认上有一定优势,我们利用这两种分析方法分析检测和鉴定出头孢克肟中的分子量为453.0的新杂质,并发现该杂质能形成一种相对稳定的二聚体杂质,并推测出该二聚体杂质的结构。
NOTES
*通讯作者。