1. 引言
合金是以二(多)元金属组成的金属材料的总称。合金储氢材料主要有四个重要系列 [1] [2]:1) AB型合金,以TiFe、Ti(Fe1-xMnx)为代表;2) AB2型Laves相储氢合金,其有三种结构,立方C15型,如MgZn2;六方C14型,如MgCu2;六方36型,如MgNi2系合金;3) AB5型稀土系合金,如LaNi5、MmNi5 (Mm为混合稀土金属)、LaNi4Cu等,以及用其它元素部分取带Ni的其它AB5合金等;4) Mg基储氢合金,如Mg88Y12、Mg-Cu等,以及添加其它少量元素的合金。
材料储氢性能的表征是多方面的 [3],但常用的储氢材料性能衡量标准是:1) 体积储氢密度,即系统单位体积内储存氢气的质量(kg/m3);2) 质量储氢密度,即系统储氢的质量与系统质量的比(%)。上述四种储氢材料之前三个重要系列中主要材料的储氢密度数据给予表1和表2。

Table 1. The volume density and quality density of different hydrogen storage materials [1]
表1. 不同储氢材料的体积密度和质量密度 [1]
Table 2. The mass and volume of required materials for storage equal-quantity (7 m3) hydrogen [1]
表2. 储存等量(7 m3)氢气所需材料的质量和体积 [1]
从表1和表2可知,合金氢化物的储氢密度是气体氢(标准状态下)的1000倍,即与1000大气压下储氢量相当;某些金属材料(如TiH2等)的储氢量仅为液态储氢量的一半。
就应用的角度而言,不仅要求材料的吸氢量,还应考虑吸氢的速度、放氢的速度、储氢的稳定性、吸氢–放氢的循环性能,以及成本和经济效益等;就新材料研发和应用基础研究而言,除应用角度要求的指标外,材料吸放氢动力学特性和热力学分析也是十分必要的,并需进一步探求影响动力学特性的根本原因和机理。
众所周知,“在哲学领域,物质决定意识;在自然科学领域,结构决定功能。”合金储氢材料的储氢性能由合金储氢材料的结构所决定。
要探求储氢材料的吸放氢机理,就必须在进行测定吸放氢动力学曲线的同时,用在线(in situ)或准动态地实时的测试分析方法观测和研究储氢材料母体的晶体结构和微结构在吸放氢过程中的演变。因此本文在总结四类合金储氢材料吸放氢动力学特性的规律(第2节)和储氢材料母体相结构在吸放氢过程中的演变规律(第3节)的基础上,通过对四类合金储氢材料的晶体结构特征及其与储氢主体(氢化物)之间的关系的研究(第4节)、氢原子在不同储氢材料中的扩散行为及热力学的综合分析,以探求合金储氢材料的吸放氢机理。
2. 典型合金储氢材料的吸氢动力学特性
研究吸放氢动力学特性的方法是在测试系统在给定压力下测定储氢材料不停顿吸氢的相对量与时间的关系曲线,即在给定氢压力情况下,测定不同温度时的吸(放)氢相对量随时间的变化(P-C-T)曲线;在给定温度下,测定不同氢压力时吸(放)氢相对量随时间的变化(T-C-T)曲线。
2.1. AB型Ti系储氢材料的吸氢动力学特性
Lee、Byun和Park等 [4] 研究了TiFe及其合金氢化动力学。图1示出TiFe在几个恒压下氢化反应分数F~时间t的作图,可见在开始阶段F随时间呈线性变化,这表示反应速率不变。直线的斜率,即反应速率随反应进程逐渐降低。上述现象表明,总的反应速率有多个梯级控制,即速率控制梯级因反应进程而变。
如果在恒压P0下,温度增加,包括活化能项的指数项增加,但平衡压力Peq或驱动力项降低。如上所述,活化能相对较小,在恒压Peq下随温度急剧变化,反应速率随温度升高而降低。因为在这个系统中不考虑驱动力项,活化能将出现负值。实事上,Stakebake [5] 已报导U氢化中负的活化能。
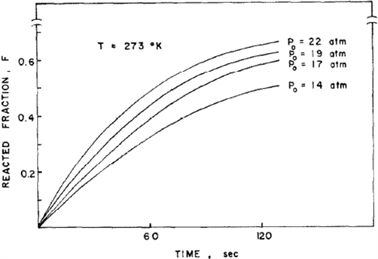
Figure 1. Hydronation dynamic curve of TiFe under 273 K and several hydrogen pressures [4]
图1. 在273 K和几种氢压下TiFe的氢化动力学曲线 [4]
Wajiad等 [6] 在不同氢压和衡压2.7 MPa下10˚C、30˚C和45˚C测定了用Cu和Y部分替代的储氢Ti-Fe-Mn合金吸氢动力学曲线(氢浓度~时间),结果表明,当纯洁的样品暴露在氢气氛的初期吸氢活性是快的;在同一温度下吸氢量随压力增加而增加;在相同压力下,吸氢量随温度升高而降低。分别10˚C 750秒,30˚C 250秒和45˚C 350秒,最大吸氢能力分别为1.67 wt%、1.41 wt%和1.32 wt%。在吸氢的潜伏期明显短,到达饱和点的溶解反应时间不超过几分钟。10˚C到达较高能力,相反,在45˚C吸氢功能相对低下,而在30˚C反应速率相对最快,到达平衡点非常快。
2.2. Laves相TiCr2储氢材料吸氢放氢动力学特征 [7] [8]
根据Ti-Cr二元相图,TiCr2有一定成分范围,而且高温时为γ-TiCr2,属六方C14结构;低温(<800˚C)为α-TiCr2,属立方C15结构;在中温段则为β-TiCr2,为C14和C15混合物。
黄太仲 [8] 为了考察不同Cr/Ti比对合金吸放氢性能的影响,设计了不同Cr/Ti比的合金,并进行了P-C-T测试,各合金在不同温度下的P-C-T曲线表明,随着测试温度的升高,合金的吸氢量降低,吸放氢间的滞后效应减弱。在273 K时,随Cr/Ti的增加,合金的滞后效应有所增强,但各合金的滞后效应还是很低的。根据各合金的P-C-T测试结果,可以得到不同温度下各合金最大吸氢量和可逆吸氢量,结果表明合金的可逆吸氢量和最大吸氢量都随温度增加而减少,而且,在273 K时,TiCr1.8时吸氢量最高,但随着温度升高吸氢量降低很快。
黄太仲等 [8] TiCr1.8-xMox (x = 0.2, 0.4, 0.6, 0.8, 1.0, 1.2)合金的P-C-T测试结果也显示上述规律,即随着测试温度的升高,合金的吸氢量降低。
2.3. AB5稀土系吸氢材料吸氢动力学特性
2.3.1. LaNi5吸氢过程动力学特性 [9]
吕曼琪、戚震中、吴平森 [9] 报导了LaNi5吸氢动力学曲线,图2给出了在相同的初压下不同温度吸氢时吸氢量随时间变化的曲线;图3给出在相同的吸氢温度,当初压不同时吸氢量与时间的关系,显然,在w~logt图上,每一条曲线都由两段斜率不同的直线构成,吸氢量高的部分所对应的那一段直线具有较小的斜率,随温度的升高或初压的降低,每一曲线上的两部分直线的斜率相差愈小,以至于十分接近。
LaNi5的吸氢反应是:
,这样的氢化物形成过程必然包括下面几个步骤:氢分子在金属表面分解为氢原子,即表面反应;氢原子向金属内部扩散;金属间化合物到其氢化物的相转变。

Figure 2. Change of LaNi5 Hydrogen Absorption quantity with time under different temperature and beginningpressure of 30.7 atm [9]
图2. 初压为30.7 atm,温度不同时LaNi5吸氢量随时间的变化 [9]

Figure3. Change of LaNi5 Hydrogen Absorption quantity with time under different beginning pressure [9]
图3. 初压不同时LaNi5吸氢量随时间的变化 [9]
2.3.2. LaNi5-xAlx储氢动力学特性 [10] [11]
文献 [10] [11] 出LaNi4.25Al0.75合金在333 K、353 K、373 K和423 K初始活化后吸氢P-C等温曲线(P-C-T),合金的储氢能力系统地降低,吸氢平台压力随温度增加而明显增加。平台压力(peq)与温度的关系函数是
。在353 K是饱和氢化物的分子式LaNi4.25Al0.75H4.2,与LaNi5H6相比较,LaNi4.25Al0.75的饱和吸氢能力降低近30%,平台压力也明显降低。
LaNi4.25Al0.75合金在1020 kPa (吸氢容器初始压力) 25个循环后吸氢的动力学曲线示d得知,在333 K和373 K 60秒内下,1个摩尔LaNi4.25Al0.75能吸氢超过4摩尔的H2,但在423 K 100秒内仅吸氢3个摩尔H2,吸氢速率随温度升高而降低。
张瑞静、吕曼祺和曹大力等研究了LaNi5-xAlx中x对储氢性能的影响 [12]。LaNi5-xAlx合金在333,353,373 K时的压力–组成等温线(P-C-T)显示,在同一温度下,Al含量增加使合金的平衡压力下降,平台长度即吸氢量有所减少。
根据上述结果,有人认为“已有研究结果普遍表明,在一定氢压条件下,合金氢化物储氢材料的饱和吸氢量随温度的升高而减小”。情况究竟如何,是不是所有合金储氢材料都是如此,且看下面的实验研究结果。
2.4 . Mg系储氢合金的吸氢–放氢动力学特征
2.4.1. Mg88Y12合金的吸氢–放氢动力学特征 [13]
Yang Yuan和Bu等 [13] 研究获得Mg88Y12合金的吸氢和放氢动力学曲线分别示于图4(a)和图4(b)。从图4(a)可知,在温度 ≥ 290˚C的试样显示较好的动力学曲线,低于290˚C,显示低劣的动力学功能,但在100˚C和150˚C下60分钟内仍能吸氢2.63 wt%和3.94 wt%;关于放氢在相对较高温度下,合金呈现好的动力学,在380˚C下放氢能10分钟内完成。但它与温度有关,在低于320˚C变得很差,如图4(b)所示,290˚C全部放氢约400分钟。
有三种模型来描述吸氢和放氢反应的不同动力学阶段:(a) 表面反应模型,(b) Johnson-Mehl-Avrami (JMA)模型,(c) 收缩体积(CV)。Yang用JAM模型拟合放氢曲线给出最好的结果,该模型用于在吸氢和放氢期间发生成核和长大过程的情况。JMA方程表示如下:
(1)
这里α是反应分数,k是速率常数,n为反应级,按照拟合结果,对放氢反应n值取3。
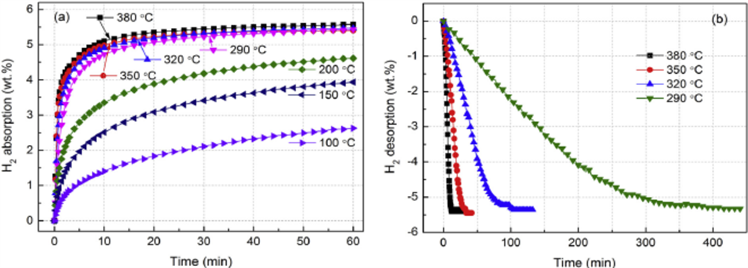
Figure 4. Dynamic curve of hydrogen abosobtion/disabsobtion for Mg88Y12 alloy under different tempreture; (a) Hydrogen absoption under 3 MPa pressure; (b) hydragen desoption lower 0.03 MPa
图4. 在不同温度下Mg88Y12吸/放氢动力学曲线。(a) 在3 MPa氢压力下吸氢,(b) 在低于0.03 MPa氢压下放氢[13]
2.4.2. Mg-Cu合金吸氢放氢动力学 [14]
Wu,Li和Zhang [14] 报导了未掺、掺Y和Ni的Mg2Cu合金在4 Mpa压力下吸氢的动力学曲线示于图5,比较可看出,未掺样品在250˚C吸氢慢,增加氢化温度到300˚C,吸氢速率变得较快,满吸氢能5分钟内完成,见图5(a);对于掺Y和Ni的样品250˚C满吸氢10分钟内完成,300˚C 2分钟内完成,见图5(b)。这意味着纳米结构的Mg2Cu合金和嵌入Mg2Cu基体中YH2/YH3对Mg2Cu非均衡反应都有好的催化效应。
为了理解未掺的Mg2Cu和掺Y和Ni样品放氢特性,等温去氢化曲线是需要的,见如图6。能够看到,在动力学上,放氢比300˚C吸氢慢。当去氢化温度增加到350˚C,Mg2Cu和掺Y和Ni样品在3分钟内分别是0.99%和1.93%的氢,说明掺Y和Ni的Mg2Cu样品显示比未掺的样品有较好的放氢动力学,

Figure 5. Time relation curve of Hydragen absobtion for hydrogen storage alloy of Mg system [14]. (a) Mg2Cu sample of no doping; (b) Sample doped Y and Ni
图5. Mg系合金储氢吸氢时间关系曲线 [14];(a) 未掺杂的Mg2Cu试样;(b) 掺Y、Ni的试样
如上所述,放氢前,在已氢化的,掺Y和Ni的MgCu2样品中,MgCu2和MgH2与Mg2NiH4,YH2及YH3一起存在,因此纳米结构的Mg2NiH4和YH2/YH3对逆反应也有催化效应。
2.4.3. 用1 mol% Nb2O5催化的Mg储氢合金吸氢放氢动力学特征 [15]
出用1 mol% Nb2O5催化的Mg储氢合金的吸氢曲线 [15],在较低氢压(0.2 Mpa)下,初始吸氢速率和饱和吸氢量都随温度的增加而增加。但氢压在1.0 Mpa下,250˚C是初始(30秒前)吸氢速率和吸氢量都低于150˚C时的情况;氢压在3.0 Mpa时,更是出现交叉情景。初始(30秒前)吸氢速率和吸氢量都低于150˚C时的情况;氢压在3.0 Mpa时,更是出现交叉情景。

Figure 6. Hydrogenation curve under equal temperature of hydragen desorbed materials; (a) Mg2Cu sample of no doping; (b) Sample doped Y and Ni
图6. 去氢化材料的等温放氢化曲线 [14],(a) 未参杂的Mg2Cu样品,(b)掺Y和NiMg2Cu样品
2.5. 小结
本节的内容总结如下:
1) 在恒定的温度和不同的氢压下,初期吸氢速率随氢压力的增加而加快,吸氢饱和量也随氢压增加而增加,这一点看是很好理解的。所述四类合金储氢材料都符合这规律;这里所说“初期吸氢速率”是初期动力学曲线斜率得知的。
2) 在恒定的氢压和不同温度下,初期吸氢速率和吸氢饱和量随温度的变化存在两种绝然相反的情况:i) 初期吸氢速率和吸氢饱和量随温度的增加而降低,AB型TiFe系、AB2型Laves相和AB5型稀土系储氢材料符合该情况,但Mg系储氢材料不符合该规律。ii) 初期吸氢速率和吸氢饱和量随温度的增加而增加,Mg系储氢材料符合这种情况,而AB型TiFe系、AB2型Laves相和AB5型稀土系储氢材料不符合;这正是本文要研究的问题。
3. 吸放氢时储氢材料的结构演变
在《储氢材料》 [16]、《金属氢化物的性质与应用》 [17] 和《Hydrogen in Intermetallic Compounds》 [18] 书籍和文献中,常常通称为“金属氢化物”储氢,顾名思义,金属及其合金在吸氢之后的产物为金属/合金的氢化合物。是不是所有的合金吸氢材料吸氢后的产物都是“氢化合物”呢?值得实验研究和分析。要研究这个问题,必须从储氢材料在吸–放氢过程中的晶体结构演变入手。
要研究储氢材料在吸放氢过程中的结构演变,关键是正确无误地对吸放氢过程中的中间产物和最终产物做出正确无误地鉴定和判断。
为下面讨论方便,引人“氢化物,(Hydride)”和“氢化固溶体(Hydrogenation Solid Solution”)两个概念。合金氢化固溶体,其晶体结构与母相相同,这个相同不仅空间群相同,晶体结构也相同。由于氢是周期表中第1号元素,原子半径远小于金属原子半径,氢原子可能仅占据母相的间隙位置,形成合金间隙式固溶体,只产生母相点阵参数的微小变化,而且这种点阵参数之变化与吸放氢浓度有一定关系。氢化物相的结构则与母相不同,因为氢原子可能不占位间隙位置,而占据一定的晶体学点阵位置,应给出不同于母相的衍射花样,因此较容易判定是否是氢化物。
如果在形成氢化物相时,氢原子占据原母相的间隙空穴位置而形成氢化物,那么该氢化物的晶体结构保持母相的结构不变,有人称之为间隙式氢化物,其本质就是氢化固溶体。
多晶物质的X射线衍射花样就如像人类的指纹一样,可做出物相的正确判断。其依据是国际衍射数据委员会(ICDD)的粉末衍射卡组(PDF)数据库——PCPDFwin [19] 中的标准数据,其方法在《物相衍射分析》 [20] 和《材料射线衍射和散射分析》 [21] 两本书的有关章节中有较详细地介绍。1) 通过对待分析样品衍射花样进行检索/匹配和判断;2) 在已知各氢化物相的晶体结构数据(晶系、空间群、点阵参数和各原子在晶胞中的位置及其占为几率)时,可对待分析花样进行Reitveld全谱拟合–精修。然而,关键的是合金储氢材料在吸放氢过程中观测的实验手段和方法。国际和国内都开发了实验室X射线源和同步辐射X射线的在线(in situ)衍射装置 [22] [23] [24],可对储氢材料在吸放氢过程作实时的动态观测和分析 [25] [26] [27];另一个重要问题是最好要有衍射分析专家的参与。
3.1. Ti系合金在吸放氢过程中的结构演变 [6]
图7(a)给出Wajid Ali等 [6] 用Cu和Y取代的刚合成Ti-Fe-Mn样品的XRD花样,由图可知,主要的衍射峰都属于具有CsCl有序结构的TiFe相,而没有发现其它相,其Rietveld分析结果见图7(b),并列入表3。
由图7和表3可知,试样吸氢前后晶体结构没有变化,仅点阵参数和晶胞体积有所增大。这表明,吸氢过程是氢化固溶体TiFe-Hn的形成,放氢过程是氢化固溶体TiFe-Hn的分解。

Figure 7. (a) XRD pattern of synthetized Ti-Fe-Mn sample substituted with Cu and Y; (b) Results of Rietveld refinement [6]
图7. (a)用Cu、Y替代合成Ti-Fe-Mn样品的XRD花样,(b) Rietveld精修结果 [6]
Table 3. Analysis results of synthetized Ti-Fe-Mn sample substituted with Cu and Y and de-hytronation sample [6]
表3. 用Cu、Y替代的Ti-Fe-Mn合成试样和已氢化样品的分析结果 [6]
3.2. Laves相TiCr2储氢合金在吸放氢过程中结构演变 [28] [29]
为了进一步研究TiCr2Laves相BCC相的吸氢本质,吸放氢前后的样品的XRD花样示于图8中。从图8可以看出,有机械合金化(MA)和机械研磨(MG)制备的两种样品都保持了BCC结构,由于氢原子进入BCC的间隙位置,导致点阵参数的增大,因此二者的谱线均向低角度方向位移。这表明这种BCC结构的吸氢方式以合金间隙式固溶体为主,并没有发生相变。由于机械合金化制备的样品吸氢量要比机械研磨的样品大,最大吸氢量达1 wt%,吸氢前后前者位移量比后者大。但在图8中机械研磨的样品衍射峰向低角度位移量反而大,这可能是因机械合金化的样品放氢量大,剩余的氢量反而比机械研磨的样品低,故后者的衍射线向低角度位移量反而大。
此外,还有表4所列AB2型Laves相的研究结果 [29]。从上述研究结果知道,Laves相储氢合金吸氢后大多结构没有变化,形成母相间隙式固溶体。
图8. 机械合金化(MA)和机械研磨(MG)两种方法制备的立方TiCr2合金吸氢前后的衍射花样 [28]
Table 4. Research resulte of structure evolution during hydrogen absorption/desorption processes for hytrogen stoarge materals of some AB2-type and Laves phase [29]
表4. 一些AB2型Laves相储氢材料吸放氢吸放氢过程结构演变的研究结果 [29]
3.3. AB5型稀土系合金在吸放氢过程中的结构演变
3.3.1. LaNi4.83Sn0.314吸放氢过程的原位X射线衍射研究 [37] [38]
袁志庆,吕光烈,曾跃武等 [37] [38] 对过计量快凝合金LaNi4.83Sn0.314吸放氢过程的原位X射线衍射研究。充放氢过程中各主要阶段的XRD图谱及Rievteld结构精修结果如图9所示。从图9中可以发现,吸氢至x = 2.0时存在两个相,α相 + β相;x = 4.0时仅存在一种相β,而放氢至x = 1.5时,又存在两个相。因此,LaNi4.83Sn0.314合金在充放氢过程中的相变呈现典型的
过程,不过该相变属膺相变,即晶体结构不变,见图9(a)和图9(c),但线条明显向低角度方向位移。α相和β同时存在,显示吸氢和放氢都有从颗粒表面到颗粒内部的过程。随着充氢过程的深入,α相的含量逐渐减小,β相的含量逐渐增加,在放氢过程中的相含量变化则与之相反。
 (a)
(a)  (b)
(b)  (c)
(c)  (d)
(d)
Figure 9. XRD patterns and Reitveld refinement results of LaNi4.83Sn0.314 alloy [37] [38]. Original sample (a), Hydrogen absorption x = 2.0 (b); x = 4.0 (c); afterward hydrogen desorption x = 1.5 (d)
图9. LaNi4.83Sn0.314合金原样(a) (x = 0.0)吸氢至x = 2.0 (b) 4.0 (c) 然后放氢至x = 1.5 (d) 时的XRD花样及Reitveld精修结果 [37] [38]
3.3.2. 镍–氢电池充放电过程准动态XRD研究 [39] [40] [41]
镍–氢电池充放电几个阶段的负极活性材料AB5的XRD图谱示于图10中。从整体来看AB5合金的结构在充电过程中没有明显的变化,但精细结构和微结构也有变化。点阵参数a和c都随充电深度增加而增加,微应变ε也随充电深度增加而增加。放电情况正好相反,但并恢复到原始状态,这表明在充–放电过程中存在某些不可逆因素。这一研究结果表明,LaNi5在镍–氢电池充放电的吸放氢过程中晶体结构保持不变,但微结构参数随之变化。换言之,吸放氢过程是LaNi5合金间隙式固溶体LaNi5-Hn的形成和分解过程。

Figure 10. XRD partters of several steps of charge (a) and discharge (b) for cathode materal AB5 alloys [39] [40]
图10. 充(a)放(b)电几个阶段的负极活性材料AB5合金的XRD图谱 [39] [40]
3.3.3. LaNi5,LaNi5-xAlx,LaNi5-xMnx和它们氘化物结构的实验研究 [42]
文献 [42] 给出LaNi5的实验中子衍射花样,粗看起来好像很不一样,但经仔细指标化和分析后发现,1) 其衍射线位置分布相同,仅发现氘化物的衍射线系统地向低角度方向位移,表明两者晶体结构相同;2) 氘化物的衍射线明显宽化,许多线条已严重重叠;3) 线条的相对强度也发生明显变化,这说明氘原子进入间隙位置后参与衍射,对衍射强度有贡献。
Endo等 [43] 以原位XRD和Rietveld全谱拟合法研究AB5型合金晶体结构的改变。吸放氢前后RTNi4.30Al0.30Mn0.40的相结构均为P6/mmm密排六方晶格,而RTNi3.55Co0.75Al0.30Mn0.40吸氢后从吸氢前的Cmmm (No.65)的正交晶格转变为P6/mmm密排六方晶格。Nakanura等 [44] 采用中子衍射技术监测合金的吸放氢过程。结果表明,LaNi5-xAlx合金的吸氢量与其Al的含量有关,随着Al含量的增加,该合金的晶体结构由P63mc (x = 0)先转变为P6mm (x = 0.1)再转变为P6/mmm (x ≥ 0.5)。这种结构变化,需进一步测定氢化物相中H原子的数目以及H的晶体学位置。
3.4. 镁系吸放氢时储氢材料的结构演变
3.4.1. Mg88Y12合金吸放氢过程的结构演变 [13]
Wu和Zhang等 [13] 研究了Mg88Y12合金吸放氢过程。图11示出刚铸造Mg88Y12合金的X射线衍射花样,能够看到,样品中存在主相Mg24Y5金属间相和少量Mg,经计算两相的相对含量分别为75.9 wt.% (Mg24Y5)和30.1 wt.% (Mg)。Mg24Y5具有α-Mn型结构,空间群I43 m,a = 1.1240 nm;Mg相为密堆六方结构,空间群P63/mmc,a = 0.3228 nm,c = 0.5222 nm,c/a = 1.6177。Mg88Y12合金中的Mg相的点阵参数比纯Mg (a = 0.3209 nm, c = 0.5211 nm)大,但c/a却比纯Mg (c/a = 1.6239)的小。上述结果表明,Y和Mg原子固溶于Mg和Mg24Y5相中。Mg88Y12合金在380˚C,3 MPa压力第一次氢化(活化)曲线示于图12(a),很清楚,第一次氢化过程是很慢的,在第一个5分钟,吸氢能力相对增加较快,然后缓慢增加,大约在550分钟后达到饱和最大氢化能力为6.479%。图12(b)显示Mg88Y12合金不同氢化阶段的XRD花样,大约在饱和吸氢能力的15%之后,Mg24Y5相的衍射峰几乎完全消失,而YH2的衍射峰出现,

Figure 11. X-ray diffraction partters of as-casting Mg88Y12 alloy [13]
图11. 刚铸造Mg88Y12合金的X射线衍射花样 [13]

Figure 12. (a) Hydrogen absoption curve of the first hydrogenation for Mg88Y12 alloy; (b) XRD partters under different hydrogen absoption steps for this alloys [13]
图12. (a) Mg88Y12合金第一次氢化吸氢曲线,(b) 合金在不同吸氢阶段XRD花样 [13]
同时Mg的衍射峰长高,进一步氢化过程能看到随Mg峰的降低出现MgH2衍射峰,并缓慢增长,当达到饱和的70%,YH3出现小的弱峰,表示少量的YH2转变成YH3,全部氢化的合金包含有MgH2,YH2和YH3相。因此,第一次氢化过程包含下述两种反应:
1) 不均衡的反应
,
2) 初期的氢化和MgH2形成:
;形成的YH2部分氢化:
Mg88Y12合金吸放氢前后的物相结构汇总于表5中,可见母合金Mg88Y12由Mg24Y5和Mg两相组成,吸氢过程Mg24Y5分解,形成YH2和YH3,Mg氢化为MgH2。
3.4.2. Mg-Cu系合金在吸放氢过程中的结构演变 [14] [15]
未掺和掺Y和Ni的Mg2Cu试样为吸氢前的XRD花样及Rietveld精修结果,并列如表6中。掺Y和Ni的Mg2Cu试样为吸放氢前的XRD花样及Rietveld精修结果,并列如表7中。
Table 5. Phase structure before and after hydrogen absoption/desorption for Mg88Y12alloy [13]
表5. Mg88Y12合金吸放氢前后的物相结构 [13]
Table 6. XRD partter and Reitveld refinement results during hydrogen absorption/desorption for no-doped Mg-Cu alloys
表6. 未掺Mg-Cu合金吸放氢时XRD花样及Rietveld精修结果
Table 7. XRD partter and Reitveld refinement results after hydrogen absorption/desorption for Mg-Cu alloys doped Y and Ni
表7. 掺Y和Ni的Mg-Cu合金吸放氢后XRD花样及Rietveld精修结果
由表6和表7可看到这样一个事实,吸氢会产生多种金属氢化物,并依靠氢化物达到储氢的目的,放氢则是氢化物分解而释放氢。至于掺Y和Ni的Mg-Cu吸氢后出现Mg2NiH4合金氢化物相有待进一步研究。
3.4.3. Mg90Ni10,Mg80Ni10Y10和Mg85Cu5Ni5Y5的在线同步辐射X射线衍射研究 [44] [45] [46]
图13示出Mg90Ni10,Mg80Ni10Y10和Mg85Cu5Ni5Y5三种样品经活化和氢化–去氢化循环11.5周期后用SR-XRD分析鉴定的结晶相,其结果列入表8。
这说明,Mg90Ni10的放氢分为下述三个步骤为:
1) Mg2NiH4去氢,即Mg2NiH4→Mg2NiH0.3。
2) 在Mg2NiH0.3存在的情况下,MgH2去氢,即MgH2→Mg去氢。
3) Mg2NiH0.3去氢,即Mg2NiH0.3→Mg2Ni。
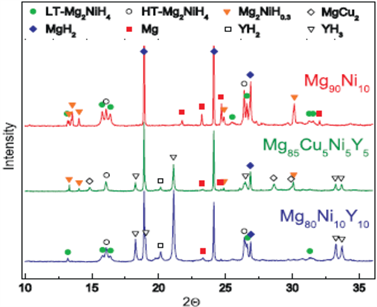
Figure 13. Synchrotron radiation x-ray diffraction partters of hytrogenated Mg90Ni10, Mg80Ni10Y10 and Mg85Cu5Ni5Y5
图13. 已氢化Mg90Ni10,Mg80Ni10Y10和Mg85Cu5Ni5Y5的同步辐射X射线衍射图谱

Figure 14. The evolution of synchrotron in situ XRD partter during equal-tempreture de-hydrogenation progress for hytrogenated Mg85Cu5Ni5Y5 under 10−2 mbar H2 presure and 200˚C
图14. 已氢化Mg85Cu5Ni5Y5合金在10−2 mbarH2压和200˚C等温去氢化过程中在线同步辐射XRD图谱的演变
图14为已氢化Mg85Cu5Ni5Y5合金在10−2 mbarH2压下和200˚C等温去氢化过程中在线同步辐射XRD图谱的演变,说明Mg85Cu5Ni5Y5去氢也分为四个步骤:
Table 8. The exist phases for three alloys after passing active-de-hytrogenation 11.5 periods
表8. 三种合金经活化和氢化-去氢化11.5周期后存在的物相
1)
2)
3)
4)
同样,在未掺和掺Cu、Y的Mg-Ni合金在吸放氢11.5周期时也出现C2/C结构的Mg2NiH4合金氢化物也值得进一步研究。
3.5. 小结
由3.1~3.4节的研究结果可总结出:Ti系、Laves相和LaNi5稀土系储氢材料在吸氢过程中,氢原子逐步进入合金间隙位置而形成固溶体,并保持晶体结构不变,储氢是依靠这种氢化固溶体来实现,即吸氢主体是合金间隙式氢化固溶体。不过多元取代的LaNi5合金情况比较复杂,发生母相结构变化。由3.4节可知,Mg系储氢材料在吸氢过程中,先是合金母相分解,然后逐步析出氢化物,储氢是依靠氢化物来实现的,换言之,吸氢的主体是金属氢化物。不过掺Y、Ni的Mg-Cu合金和掺Y、Cu的Mg-Ni合金吸氢后除MgH2、YH2、YH3外,还出现Mg2NiH4合金氢化物到是例外,值得进一步研究。
Mg2Ni属六方结构,P6222,No.188空间群,Mg2NiH4属单斜结构,c2/c,No.15空间群,那么Mg2Ni的氢化过程为Mg2Ni→Mg2NiH0.3→Mg2NiH4。在Mg-Ni二元相图中查得有Mg2Ni和MgNi2两种中间相存在,其分别含Ni的重量百分数约为55%和83%。建议最好是配置Mg2Ni单相合金试样,进行吸放氢试验,并跟踪测定母相和中间产物以及满吸氢后的最后产物的物相结构和点阵参数,就能判断上述结果是否正确,是否存在合金氢化物Mg2NiH4。
4. 典型储氢合金材料母体和氢化物的晶体结构特征
4.1. 典型密堆金属点阵的空间利用率、间隙位置的大小和数目 [47]
在金属中有三种堆积结构,镁、铍的晶体是密堆六方(CPH),铜、银和金是面心立方(FCC)密堆,第三种普通堆积类型是体心立方(BCC)堆积,它们存在于K、Na和β-Ti金属中。三种密堆金属一些重要结构参数和空间利用率(%)的计算结构列入表9中。
4.2. TiFe的母体结构和氢化物结构特征
研究得最多的Ti系合金是TiFe,属CsCl型结构。有人认为TiFe合金的氢化行为是经历TiFe (立方晶)至TiFeH1.04 (四方晶,中间氢化物相)再至TiFeH1.95 (立方晶,饱和氢化物相)的过程。
Table 9. Same importantive structure parameters of three kinds of dense-stacking metals
表9. 三种密堆金属一些重要结构参数 [47]
在网络上输入“TiFe储氢合金的氢化物”关键词,也发现有TiFeH1.04和TiFeH1.95的说法,这些资料的可信度如何?首先,CsCl型结构的TiFe在吸放氢过程中的氢化物是TiFeH1.04和TiFeH1.95,但缺乏实验证据,PCPDFwin数据库中查不到这种化合物的卡片。然而,Wajid,Ali等 [6] 用Cu和Y取代的Ti-Fe-Mn合金吸氢后仍保持有序BCC结构。从表8可知,BCC结构单胞中有8个四面体间隙和6个八面体间隙,其间隙尺度分别为0.291R和0.154R,Ti的原子半径为0.145 nm,0.145 nm × 0.291 = 0.0423 nm,因Fe的原子半径比Ti小,仅为0.135 nm,实际间隙半径大于0.0423 nm,故四面体间隙能容纳半径为0.045 nm的H原子。
4.3. Laves相储氢合金的晶体结构和间隙位置
C15型以MgZn2为典型,立方结构,原子面的堆垛顺序为ABCABC,空间群Fd
3m
;C14型以MgCu2为典型,六方结构,原子面的堆垛顺序为ABABAB,空间群为P63/mmc;C36型以MgNi2为典型,六方结构,原子面的堆垛顺序为ABACABAC,空间群为P63/mmc。三种Laves相储氢合金的晶体结构参数和间隙位置给予表10中。
根据硬球堆积模型,形成Laves相的理想原子半径比(rA/rB)为1.225。但在许多二元或似二元Laves相合金中,rA、rB金属原子半径之比均偏离1.225,该半径之比可以在1.05~1.68范围内变化。除原子尺寸因素外,Laves相结构还取决于电子密度因素。统计结果表明,Laves相结构的电子浓度(价电子/原子)为:C14型为2.0;C15型为1.7;C36型为1.90。
Laves相合金的所有晶胞间隙均为四面体间隙。该四面体间隙有三种类型,即A2B2、AB3和B4;每单位晶胞的四面体间隙总数为17,其中A2B2为12,AB3为4,B4为l。当Laves相合金吸氢时,氢占据四面体间隙,使晶胞体积膨胀,但晶型则不发生变化。在Laves相合金氢化固溶体中,并不是所有的四面体间隙都被氢原子占据,而是部分间隙可被氢原子占据,这主要是由于静电限制造成。
Table 10. The crystal sytucture paramers and interstitial hole of three hydrogen storge alloys with AB2 type Laves phase [29]
表10. 三种AB2型Laves相储氢合金的晶体结构参数和结构中的间隙空隙 [29]
4.4. LaNi5合金的母体结构和氢化物结构特征 [48] [49] [50]
4.4.1. Magee和Liu对LaNi5相储氢合金的研究结果 [48]
LaNi5晶体属CaCu5型结构,属六方晶系,空间群为P6/mmm (No.191)。早在1981年,Magee和Liu等 [48] 就以“金属间化合物结构与氢化物形成之间的关系”为题发表了四大家族金属间化合物四面体间隙空穴的数目、大小、位置和对称性的研究结果。表11给出LaNi5位置。
4.4.2. LaNi5Hx<6.0间隙空穴
LaNi5吸氢后晶系及空间群不变,晶胞发生膨胀,晶格常数改变较大,吸氢前后晶胞参数见表12。
晶格中的空隙可以是由同种金属原子构成,也可以是由不同金属原子混合构成,因而有多种类型,其中最主要的是四面体空隙和八面体空隙。具体到一个LaNi5晶胞中,共有五种类型空隙37个,即6 m型空隙6个,12n型空隙12个12o型空隙12个,4 h型空隙4个,
3f
型空隙3个。图15标明了LaNi5晶胞中五种空隙的立体示意图。图16给出氢化物β相五位置模型中间隙位置。
Table 11. The interstitial hole in structure of AB5 intermetal compouds [57]
表11. AB5金属间化合物结构中的间隙空穴 [57]
Table 12. Change of lattice parameter and crystal cell volume before and after hydrogen absoption for LaNi5
表12. LaNi5吸氢前后的点阵参数及晶胞体积的变化

Figure 15. The sketch map of five interstitial seats in LaNi5
图15. LaNi5种五种间隙位置示意图

Figure 16. The interstitial seats in five seat model for hydrid β phase
图16. 氢化物β相五位置模型中间隙位置
Cao和Chen等 [10] 则认为主要有四面体、六面体、八面体和十二面体四种间隙位置,其重要参数如表13所示,表12中也列出氢化物β相五位置中间隙位置(见图16)。
Table 13. The interstitial seats in LaNi5 structure [50]
表13. LaNi5结构中的间隙位置 [50]
王宏,刘祖岩 [50] 在《LaNi5最大储氢量的晶体学分析》中根据氢的原子半径为0.045 nm (或0.078 nm),所以理论上讲一个LaNi5晶胞能容纳18个氢原子,一个晶胞中有一个La原子和5个Ni原子,即可计算最大储氢的质量分数1.379%,这与实验测得的储氢质量分数为1.35~1.38%基本相符 [49] [50]。
文献 [51] [52] [53] [54] [55] 也对LaNi5型储氢材料吸放氢主体的晶体结构和微结构也作了研究。
4.5. Mg基储氢合金母体结构和氢化物结构
Mg88Y12的母相为Mg24Y5和Mg,氢化物是MgH2、YH2和YH3,显然是Mg24Y5分解后形成的。未掺Y、Ni的Mg2Cu的母相是Mg2Cu和MgCu2,吸氢后的产物是MgCu2和MgH2,只有MgH2是氢化物,说明Mg2Cu分解为MgCu2和Mg,Mg与H形成氢化物;而掺Y和Ni的合金母相为Mg2(Cu, Ni)和YH,吸氢后形成氢化物有MgH2、Mg2NiH4、YH3和YH2,说明Mg2(Cu, Ni)已经分解。
至于MgH2、Mg2NiH4、YH3和YH2等氢化物相是怎样形成的讨论如下。存在两种可能:1) 母相分解产物Mg和Y在没有形成结晶相时的原子态与H反应而形成MgH2、YH2和YH3;2) 母相分解产物Mg和Y在已形成结晶相后与H反应而形成。因为Mg和Y都属A3型密堆六方结构,空间群P63/mmc,No.194,单胞中有两个原子,其位置分别是:0,0,0;1/3,2/3,1/2。而MgH2属P42/mnm,No.136,YH2和YH3分别属Fm
-3m
,No.225和P
-3c
1,No.165,它们属不同结构类型,可见第1种可能性比较大。
Mg2Ni是Mg2(Cu,Ni)相的分解产物,属六方结构,P6222空间群,No.180,而Mg2NiH4为单斜晶系,C2/c空间群,No.15,Mg2NiH4则是固–气反应的产物,属第2种情况。
5. 合金储氢材料吸放氢动力学机理和热力学分析
本节是综合分析第2~4节所描述的内容的基础上,讨论储氢材料的吸氢、放氢和储氢的机理。
5.1. 表面反应——吸附和脱附 [3] [56] 和H在储氢材料中扩散
氢分子和离解后氢原子与储氢材料表面接触就会发生反应,包括物理吸附、化学吸附,然后渗透到材料表面等。此外也存在脱附现象。这种吸附和脱附与吸氢时的氢压、温度有关,还与吸氢材料种类有关。
初期到达材料表面的氢原子通过各种路径向内部扩散。一般来讲,有沿晶界和晶内扩散。从第4节介绍知道,不同密堆结构的空间利用率不同,间隙位置数目和间隙空间的大小不同,这严重影响H在储氢合金的晶内扩散。还有储氢材料中晶体缺陷多少和分布也会影响H在储氢合金的晶内扩散。如果晶粒细小,晶界就多,扩散速度相对较快,因此纳米化材料有较好的扩散性能。
5.2. 合金储氢材料的储氢机理
5.2.1. 一般描述
一般认为,金属或合金(用M代表)与氢作用可以生成金属(合金)氢化物(MHn)。其反应方程式为:
(生成热)
该反应是一个可逆过程,正向反应时,金属材料吸氢,并放出热量;逆向反应时,金属氢化物释氢,吸收热量。这样,只需要改变温度与压力,就能使反应向正向或逆向反复进行。达到金属(合金)储氢或释氢的目的。
储氢过程的能量变化可包括三部分:1) 氢气由气态分子离解成原子状态需要能量ΔH1,即氢的离解−4.75 eV;2) 氢进入金属晶体后与金属发生作用,即健合所产生的能量变化ΔH2,
因此有
这一步,无论是对什么储氢材料,在同样温度和氢气压下,可认为是一样的。问题是
这一步,由于M的种类繁多,对于不同的合金储氢材料差异非常大,甚至还可能分成许多不同的小的步骤。M是什么?MH6.5是什么?都需要进行实验测定和研究。
5.2.2. 氢原子进入母相的间隙位置——氢化固溶体的形成
从对TiFe合金、LaNi5和Laves相三类吸氢材料母相结构的间隙空穴的数量、大小和位置的讨论中,当间隙空穴半径大于或等于氢原子半径时,氢原子进入合金储氢材料的主体点阵的间隙位置,由少到多,直至饱和,但合金储氢材料主体的晶体结构不变,不过其微结构、精细结构会发生变化,这就是氢化固溶体的形成过程。
如此看来,把LaNiH6.5看成合金氢化物,显然是不妥的,而是氢化固溶体,通式写成LaNi-Hn更为科学合理,其中的n在吸氢的不同阶段n值不同,从4.2节知道,n的最大理论可达18。放氢则是可逆过程,即氢化固溶体逐步分解而释氢。
5.2.3. 储氢材料母相的分解和氢化物的析出和形成
就合金储氢材料也是多种多样的,它们晶体结构、成分、组成不同。从第2、3节的介绍可知,合金储氢材料可能单相的,也可能是多相的,它们的晶体结构也会不同。比如Mg88Y12由具有α-Mn型结构,空间群I
43m
,a = 1.1240 nm的Mg24Y5和具有密堆六方的Mg组成,从第3.4和4.4节介绍吸放氢过程Mg系储氢材料的结构演变的XRD分析结果知道,Mg88Y12合金在380˚C,3 MPa压力第一次氢化(活化),在第一个5分钟,吸氢量相对增加较快,然后缓慢增加,大约在550分钟后达到饱和最大氢化能力为6.479%。图12(b)显示Mg88Y12合金不同氢化阶段的XRD花样,大约在饱和吸氢能力的15%之后,Mg24Y5相的衍射峰几乎完全消失,而YH2的衍射峰出现,同时Mg的衍射峰长高,进一步氢化过程能看到随Mg峰的降低出现MgH2衍射峰,并缓慢增长,当达到饱和的70%,YH3出现小的弱峰,表示少量的YH2转变成YH3,全部氢化的合金包含有MgH2,YH2和YH3相的形成。现写出:吸氢45%时,
很明显,储氢材料之母相Mg24Y5分解消失,析出储氢相YH2和YH3,但Mg相还存在,而100%吸氢后
母相全部消失,吸氢相全部形成,因此储氢材料吸氢就伴随着母相分解和氢化物相的析出/氢化物相的形成过程,这个过程包含了新相成核和长大过程。放氢过程则是氢化物储氢相逐步分解而释氢。这明显表明,不能把合金Mg88Y12氢化物写成Mg88Y12Hn,因为Mg88Y12合金包括
两种相,同样也不能把氢化物写成
或
,而有一个过程,即
。
同理,对于Mg-Cu系,
母相 % 吸氢后主要相 % 放氢后的主要相 %
Mg2Cu 83 MgCu2 76 Mg2Cu 82
MgCu2 17 MgH2 27 MgCu2 16
可见,母相Mg2Cu分解,MgH2形成,至于母相中的MgCu2,其为Fd
3m
结构的Laves相,在吸放氢过程是否形成氢化固溶体MgCu2-Hn还有待进一步研究,其方法就是配置单相MgCu2的试样,进行吸放氢试验,同时跟踪吸放氢过程测定母相,中间相和最终相的晶体结构及精确测定点阵参数,便能做出最后的判断。
对于掺Y、Ni的Mg-Cu合金和掺Y、Cu的Mg-Ni合金吸氢后除MgH2、YH2、YH3外,还出现Mg2NiH4合金氢化物到是例外。
Mg2Ni属六方结构,P6222,No.188空间群,Mg2NiH4属单斜结构,c2/c,No.15空间群,那么Mg2Ni的氢化过程为
。在Mg-Ni二元相图中查得有Mg2Ni和MgNi2两种中间相存在,其分别含Ni的重量百分数约为55%和83%。建议最好是配置Mg2Ni单相合金试样,进行吸放氢试验,并跟踪测定母相和中间产物以及满吸氢后的最后产物的物相结构和点阵参数,就能判断上述结果是否正确,是否存在合金氢化物Mg2NiH4。
5.3. 吸放氢机理的热力学分析
张秀兰,黄整,陈波等 [57] 已对LiNi5储氢过程做了热力学分析。根据平衡状态下的热力学函数,导出了能够完整描述整个实验范围P-C-T关系平衡公式,利用P-C-T平衡公式对不同温度下平衡压力与储氢量的变化曲线进行了拟合分析,计算讨论了储氢过程平衡反应的热力学函数。下面仅作定性的讨论。
从合金储氢材料的晶体结构特性来考虑可分为两类:1) 晶体结构为密堆或接近理想密堆的,容易与H形成氢化物,如纯Mg最接近理想密堆(c/a = 1.633);2) 第二类是原子在点阵空间利用较低,存在许多间隙位置,而且间隙空间的半径大于H原子半径,容易形成氢化固溶体。
从合金储氢材料真正储氢主体来考虑也可分为两类:1) 一类是氢化物,换言之,吸氢过程是母相分解和继后氢化物析出形成,放氢过程是氢化物的分解过程;2) 一类是氢化固溶体,即吸氢过程是H原子扩散嵌入储氢材料晶体的间隙空穴位置,逐步形成含氢稀固溶体,直至形成饱和/或过饱和固溶体,放氢则是氢化固溶体的分解而释氢。
储氢主体为属氢化物这一类与氢化固溶体一类相比较,在扩散机制、储氢主体的形成过程存在很大差异。
前者氢原子主要沿晶界扩散,母相分解和新相形成优先在晶界上发生,然后扩展到晶粒内部;而后者则主要是向晶粒内部扩散,进入储氢材料母相的间隙位置,氢化固溶体形成也是晶粒内部开始。
两类储氢主体的形成过程不同,它们的热力学过程也应有明显差别。比如,沿晶界扩散需要的能量较低,而嵌入晶粒内的间隙位置需克服较高的势垒;相对而言,氢化物的成核和长大花的能量较小,而氢化固溶体的形成要花较大的能量。
在一定的氢压下,温度升高,母相更容易分解,氢化物更容易形成,因此该种储氢材料的吸放氢动力学特征显示吸氢速度和吸氢量随温度升高而增加;相反,温度升高对氢原子扩散进入合金母体点阵影响不大,故储氢主体为氢化固溶体的储氢材料的吸氢动力学特性表现为吸氢速度和吸氢量随温度升高而有所降低。
6. 总结、展望和建议
6.1. 总结
通过上述2~5节的介绍和以上综合分析可总结合金储氢材料的吸/放氢动力学特性如下:
1) 在较低温度下,不同氢压时两类材料的吸氢动力学曲线显示出初始吸氢速率和饱和吸氢量都随氢压的增加而增加;但当温度过高,会出现以氢化物储氢的储氢材料仍显示上述情况,而以氢化固溶体储氢的储氢材料,可能因过高温度严重影响氢化固溶体的放热效应,不会完全显示上述情况;
2) 在较低的氢压下,不同温度时的吸氢动力学曲线,以氢化物为储氢主体的储氢材料显示出初始吸氢速率和饱和吸氢量都随温度的增加而增加,而以氢化固溶体储氢的材料则显示初始吸氢速率和饱和吸氢量都随温度的增加而降低,这是因为温度对氢化物和氢化固溶体的形成影响不同而引起。同样,当氢压过高时,两类储氢材料都可能出现交叉情景。比如用1 mol%Nb2O5催化的Mg储氢合金的吸氢曲线 [14],在较低氢压(0.2 Mpa)下,初始吸氢速率和饱和吸氢量都随温度的增加而增加。但氢压在1.0 Mpa下,250˚C是初始(30秒前)吸氢速率和吸氢量都低于150˚C时的情况;氢压在3.0 Mpa时,更是出现交叉情景。
6.2. 展望和建议
关于金属储氢研究进展已有不少新的报道 [58] [59] [60],但多注意材料本身的研究。随着新型合金储氢材料的出现,分别以氢化物为储氢主体和以氢化固溶体为储氢主体的两类合金材料都会有新的发展,可能出现两者兼存的混合型储氢材料,就如像前述的Mg-Cu储氢合金那样,其中Mg2Cu相分解,继后形成MgH3,而MgCu2相属Laves相,在吸氢过程中可能形成氢化固溶体。
此外,还可能出现以吸附氢为主体的储氢材料,如纳米/介孔储氢材料 [61]。
建议对新研发的储氢材料要注意开展以下几方面的测试分析研究工作:
1) 开展对原始储氢材料存在物相鉴定,分析研究这些物相的晶体结构特征,在PCPDFWIN的数据库查找是否存在相关的氢化物及其衍射数据;
2) 在研究材料吸–放氢动力学和热力学时,要特别注意研究储氢材料原始物相在吸–放氢过程中的结构演变及微结构的变化。从第3节所介绍的XRD分析实验可知,XRD分析试样未做什么特殊处理。看来当能忽略表面吸附对储氢贡献时,不一定需要作在线(in situ)的XRD分析,可在吸–放氢的各个阶段取样,因为氢化物和氢化固溶体都是相当稳定的;当实验结果显示无相变出现时,要特别注意其衍射线条的位移、宽化现象和线条相对强度的变化,并作进一步分析。在进行这种测试分析时,有条件的话,最好使用中子衍射技术,因为H和金属的X射线的散射因子相差很大,H原子对X射线衍射强度的贡献甚少。而H和金属对中子射线的散射因子相差较小,较能显示H原子对衍射强度的贡献。
3) 吸氢–放氢的循环性能是十分重要的,美国能源部2015年储氢目标规定应具备1500周期吸放氢循环的能力。在研究循环性能及循环性能衰减的同时,注意测试分析研究储氢材料在满吸态和全放态试样的晶体结构及微结构随循环周期的变化,以探索循环性能衰减的原因,为探索提高合金储氢材料的循环性能提供思路。
上述建议看来对已研发/已应用储氢材料也是需要的,特别是对典型的储氢材料开展系列研究还是很有必要的。