1. 引言
双醋瑞因 [1] 可通过抑制成骨性细胞RANKL的表达或抑制其上游调控因子而抑制由L-1B刺激的骨吸收,从根本上治疗退行性关节炎等疾病。同时可诱导软骨生成,具有止痛、抗炎及退热作用,不抑制前列腺素合成,对骨关节炎有延缓疾病进程的作用 [2]。有文献报道双醋瑞因的相关合成路径 [3] - [8],但是没有两个已知单乙酰化(4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1a);5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1b))杂质的合成报道,有关报道也只是采用制备液相分离鉴别 [9] [10],也没有很好地得到产物,即使采用制备液相来分离纯化,成本高昂;并且该化合物不稳定,极易水解。在仿制药的申报及生产过程中需要相关杂质作为对照品 [11],然而国内目前的销售价格在100 mg/2万元左右,因此两个已知杂质会给双醋瑞因化学仿制药的申报带来高昂的成本,研究一条能制备出该杂质的合成工艺很有意义。
基于(1a)、(1b)化合物是一个平面结构,溶解性很差 [12],很难直接通过层析硅胶柱分离纯化,因此我们从改变功能团改变其理化性质的基本原理出发,用溴苄增强羟基和羧基的酯溶性,硅胶柱层析分离纯化,再使用10%的Pd/C催化氢解。本研究以芦荟大黄素为起始原料,经氧化、乙酰化、溴苄保护、钯炭催化还原4步反应,得到双醋瑞因的两个关键杂质(1a)、(1b),合成路径见图1。经ESI-MS,IR、1H NMR、13C NMR、元素分析确证。
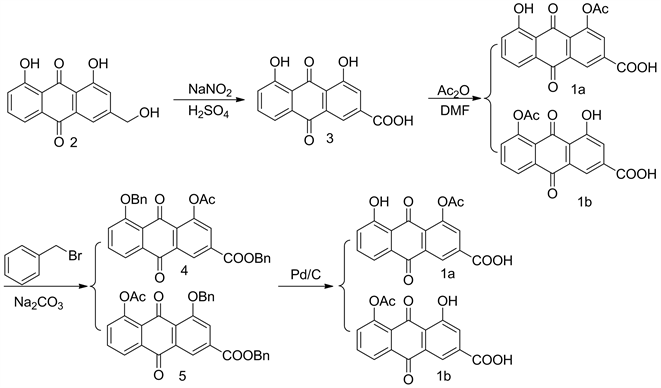
Figure 1. The synthetic route of the title compound
图1. 合成路线图
2. 实验部分
2.1. 主要仪器与试剂
Varian Mercury-400核磁共振仪(美国Varian公司);Vario EL元素分析仪(德国Elementar元素分析系统公司);Magna FT-IR-750光谱仪(美国Nicolet公司);1100型高效液相色谱仪(配有二级管阵列(DAD)检测器,美国Agilent公司);Xevo G2-XS QTof高分辨质谱仪(美国Waters公司);所用试剂均为分析纯。
2.2. 合成
2.2.1. 大黄酸(3)的合成
在250 mL三口烧瓶中加入硫酸120 mL,缓慢加入亚硝酸钠25.55 g (370.40 mmo1),加完升温至120℃后并保温于120℃缓慢加入10.0 g (37.04 mmo1)芦荟大黄素2,加完保温120℃继续搅拌反应4小时;降至室温,将反应液缓慢倒入搅拌的1000 mL冰水中,继续搅拌0.5小时,抽滤,滤饼用水2 × 100 mL洗涤,抽至无液体流出,滤饼105℃干燥,得粉状固体10.0 g,加入N,N-二甲基甲酰胺100 mL加热溶解澄清后,室温放置析晶12小时,抽滤,滤饼用水2 × 40 mL洗,抽至无液体流出,滤饼105℃干燥,得黄色粉末状结晶9.6 g,收率:91.25%。mp 316℃~318℃,文献 [3] (317℃~319℃),1H NMR (400 MHZ, DMSO-d6),δ = 7.42 (dd, J = 1.2 Hz, 8.4 Hz, 1H, Ar-H),7.75~7.77 (m, 2H, Ar-H),7.84 (dd, J = 7.5 Hz, 8.3 Hz, 1H, Ar-H),8.16 (d, J = 1.2 Hz, 1H, Ar-H),11.93 (s, 2H, OH),13.70 (s, 1H, COOH);IRν:3061,2604,1692,1629,1608,1570,1452,1266,1188,1075;ESI-MS m/z:Calcd for C15H8O6Na {[M-H]−}284.032 1,found 284.0323。
2.2.2. 单乙酰化混合物(1a, 1b)的合成
于500 mL圆底瓶中加入9 g (31.69 mmol)化合物3,乙酸酐7 mL (74.05 mmol),1,4-二氧六环90 mL,加完油浴回流反应20小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V (乙酸) = 20:10:1]。降至60℃减压蒸出大量1,4-二氧六环,加入200 mL水搅拌0.5小时,抽滤,滤饼用50 mL水淋洗一次,抽至无液体流出,滤饼50℃减压蒸干得黄色粉状固体9.5 g,该混合固体未经纯化直接往下合成。
2.2.3. 4-乙酰基-5-苄氧基-9,10-二氧-9,10-二氢蒽-2-羧酸苄酯(4)与5-乙酰基-4-苄氧基-9,10-二氧-9,10-二氢蒽-2-羧酸羧酸苄酯(5)的合成
将混合固体9 g加入到250 mL圆底瓶中,再加入N,N-二甲基乙酰胺90 mL,无水碳酸钠5.8 g,溴化苄4.18 mL,加完升温至100℃搅拌反应4小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯) = 2:1]几乎无混合物(1a, 1b)点。停止加温,降至室温加入200 mL CH2Cl2,冰水400 mL,加完搅拌15 min,分出CH2Cl2,CH2Cl2层2 × 200 mL水洗,CH2Cl2无水硫酸钠干燥,40℃减压回收CH2Cl2得浸膏状产物,加入100 mL石油醚有红棕色固体析出,搅拌1小时,抽滤,滤饼25 mL石油醚淋洗一次抽干,滤饼45 ℃减压干燥,得红棕色混合粉状固体11 g。
将11 g红棕色粉状固体溶于110 mL乙酸乙酯中,加入22 g硅胶拌样干法上样过硅胶层析柱,[洗脱液,V (石油醚):V (乙酸乙酯) = 3:1)]洗脱液分别减压蒸干得化合物4 2.6 g,ESI-MS m/z:Calcd for C31H22O7Na {[M-H]−} 505.1366,found 505.1367;化合物5 1.2 g,ESI-MS m/z:Calcd for C31H22O7Na {[M-H]−} 505.1366,found 505.1369。
2.2.4. 4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸 (1a)的合成
将2.5 g化合物4加入到50 mL圆底瓶中,加入THF 25 mL,10%的Pd/C 0.25 g,加完氢气真空切换3次,后通氢气室温搅拌反应12小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V (乙酸) = 20:10:1]。抽滤,滤液40℃减压蒸干得浅红棕色固体1.6 g,加入乙酸乙酯16 mL搅拌1小时后抽滤,滤饼5 mL乙酸乙酯淋洗一次,抽干,滤饼45℃减压干燥,得浅红棕色粉状固体1.2 g。收率:75.16%,mp:246℃~247℃;1H NMR (400 MHZ, DMSO-d6),δ = 2.42 (3H, S, CH3),7.35 (1H, d, J = 8.4 Hz, Ar-H),7.67 (1H, d, J = 7.2 Hz, Ar-H),7.77 (1H, t, J = 8.0 Hz, Ar-H),8.01 (1H, d, J = 1.6 Hz, Ar-H),8.54 (1H, d, J= 1.6 Hz, Ar-H),12.17 (1H, s, OH)。13C NMR (100 MHZDMSO-d6):δ = 20.67,116.40,118.91,124.45,125.11,126.82,130.14,132.33,134.99,137.04,137.07,150.17,161.37,164.75,168.82,180.44,186.58;IRν:3445,3074,1770,1691,1640,1609,1578,1459,1419,1372,1353,1331,1312,1278,1245,1225,1193,1155,1097,1075,1051,1021;ESI-MS m/z:Calcd for C17H10O7Na {[M-H]−}325.0427,found 325.042 6;Anal calcd for C17H10O7:C 62.34,H 3.32,O 34.46;found C 62.58,H 3.09,O 34.33。
2.2.5. 5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1b)的合成
将1.2 g化合物5加入到50 mL圆底瓶中,加入THF 12 mL,10%的Pd/C 0.12 g,加完氢气真空切换3次,后通氢气室温搅拌反应12小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V(乙酸) = 20:10:1]。抽滤,滤液40℃减压蒸干得浅红棕色固体0.7 g,加入乙酸乙酯10 mL搅拌1小时后抽滤,滤饼5 mL乙酸乙酯淋洗一次,抽干,滤饼45℃减压干燥,得浅红棕色粉状固体0.5 g。收率:69.44%,mp:238℃~239℃;1H NMR (400 MHZ, DMSO-d6):δ = 2.41 (3H, S, CH3),7.60 (1H,d, J = 8.0 Hz, Ar-H),7.71 (1H, d, J = 7.2 Hz, Ar-H),7.96 (1H, d, J = 8.0 Hz, Ar-H),8.08 (1H, d, J = 1.2 Hz, Ar-H),8.13 (1H, d, J = 1.2 Hz, Ar-H),12.14 (1H, s, OH) 13CNMR (100 MHZ, DMSO-d6) δ = 20.67,118.39,118.67,124.02,124.13,125.21,130.56,132.85,134.53,136.14,137.94,150.01,161.06,165.15,168.80,180.60,186.77;IRν:3442,3096,2989,2853,2608,2545,1764,1705,1677,1645,1591,1567,1484,1445,1417,1368,1339,1270,1236,1205,1193,1155,1094,1045,1020,996。ESI-MS m/z:Calcd for C17H10O7Na{[M-H]−} 325.0427,found 325.0426;Anal calcd for C17H10O7:C 62.19,H 3.32,O 34.34;found C 62.58,H 3.09,O 34.33。
3. 结果与讨论
通过相关实验验证,本研究很容易得到双醋瑞因的两个关键杂质4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸和5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸;在混合物(1a, 1b)的制备过程中,主要产物为二乙酰大磺酸和(1a, 1b),其HPLC见图2。
[HPLC面积归一化法,色谱柱:用十八烷基键合硅胶(ZORBAX SB-C18)为填充剂(4.6 mm × 250 mm, 5 μm);流动相:甲醇-乙腈-0.005 mol/L磷酸二氢钾,用磷酸溶液调节至pH 1.6 (38:10:52);检测波长为254 nm;柱温为35℃;流速为1.0 mL/min。]
由于该混合物的化学结构决定其化学性质,并且1a、1b只是位置不一样,理化性质相似,溶解性也很差,在改变功能团来改变其理化性质的过程中使用过三甲基硅醚、叔丁基二苯基硅醚、叔丁醇和溴苄、对甲氧基溴苄、三苯甲基醚,通过实验验证前三个试剂保护时脱保护采用酸水解其酯建也会断裂,后三个保护试剂比较后溴苄效果最好。但是在用10%的Pd/C催化氢解制备1a、1b时,所使用的溶剂THF一定为无水,通过实验得出普通THF作为溶剂时产物为大黄酸3。
基金项目
贵州省化学药物开发利用工程实验室;贵州省普通高等学校药物化学工程研究中心(黔教合KY字[2014]219号)。