1. 引言
布洛芬(Ibuprofen, IBU),又名异丁苯丙酸,是一种非甾体类抗炎药,具有解热镇痛、抗炎等作用 [1]。其通过抑制环氧合酶的活性使前列腺素等炎症介质合成减少 [2],从而达到镇痛抗炎效果,临床上已证明布洛芬具有治疗感冒和轻微发热的作用,且退热效果比同类药物更为明显,故IBU是世界卫生组织(WHO)和美国药品监督局(FDA)唯一推荐的儿童退烧和抗炎的首选药 [3]。
IBU的水溶性低,在水中溶解度约为60 mg/L,且在体内溶出缓慢,生物利用度低,因此难溶性药物的增溶技术已成为药剂学的研究热点 [4]。其中固体分散技术因其操作简单,增溶效果较好,被广泛的应用。长期以来有报道利用交联聚维酮(PVPP)和聚乙二醇(PEG6000)等将难溶性药物制成固体分散体,显著增加了药物的溶出度。
本文以布洛芬为原料药,体外溶出结果为指标,比较溶剂法、熔融法在不同制备工艺参数下的溶出度,从而得出最佳制备条件,并对固体分散体(Solid Dispersion, SD)进行物相鉴别分析。
2. 实验仪器与材料
2.1. 仪器
UV-5500PC紫外分光光度计(上海元析仪器有限公司);HH-4数显恒温水浴锅(常州澳华仪器有限公司);RC806D溶出度仪(天津市天大天发科技有限公司);85-1A磁力搅拌器(巩义市予华仪器有限责任公司);电子分析天平(上海卓精);智能型电热恒温鼓风干燥箱(上海琅玕实验设备有限公司);旋转蒸发仪器(瑞士BUCHI公司);Q2000-差示扫描量热仪(美国TA公司);X′PertPowder X射线衍射仪(荷兰帕纳科公司);ΣIGMA+X-Max20电子扫描显微镜能谱仪(德国蔡司公司)。
2.2. 材料
布洛芬原料药;聚乙二醇6000 (天津市科密欧化学试剂有限公司);聚乙烯吡咯烷酮(深圳市优普惠药品股份有限公司);无水乙醇为分析纯(川东化工)。
3. 方法与结果
3.1. 布洛芬体外分析方法的建立
3.1.1. 含量测定
1) 吸收波长
精密称定0.024 g IBU于15 ml容量瓶,加适量无水乙醇溶解并定容至刻度线。以无水乙醇为空白对照,在200~400 nm进行全波长扫描,结果表明,IBU在264 nm处有最大吸收,故确定方法的测定波长为264 nm。
2) 储备液的制备
精密称定0.2030 g IBU于15 mL的容量瓶中,加少量无水乙醇溶解,定容至容量瓶的刻度线作为储备液。
3) 标准曲线的制备
分别精密量取“3.1.1.2”项下储备液0.5、1、2、3、4、5、6 mL于15 mL的容量瓶中,用无水乙醇定容至刻度,得0.0406,0.0812,0.1624,0.2436,0.3248,0.4000,0.4872 mg/l系列浓度对照品溶液,以无水乙醇作为空白对照。在最大吸收波长264 nm测定其吸光度,以吸光度(A)为纵坐标,以IBU对照品浓度(C)为横坐标,得标准曲线方程为:A = 1.4209C − 0.0015 (r = 0.9996),结果表明IBU在0.0406~0.4872 mg/ml线性关系良好。
4) 重复性实验
精密量取3 ml储备液6份分别置于15 ml容量瓶,无水乙醇定容至刻度线,进样测定。得IBU的相对标准偏差(RSD, relative standard deviation)为0.91%,表明分析方法重现性良好。
5) 精密度实验
精密量取“3.1.1.2”项下储备液0.5、3、6 mL分别置于15 mL容量瓶中,用无水乙醇定容至刻度线,配制成3种不同浓度(0.0406、0.2436、0.4872 mg/mL)的布洛芬溶液。在1 d内各浓度样品连续进样3次,得不同浓度日内RSD分别为0.91%,0.35%,0.17%;每天测一次,连续测定5 d,得低、中、高浓度日间RSD分别为1.16%,0.84%,0.41%;结果表明日内RSD和日间RSD良好,符合含量测定要求。
6) 样品含量测定
精密称定IBU固体分散体(相当于药品0.1 g),置于15 ml的容量瓶中,加适量无水乙醇溶解,并定容至刻度线,在波长264 nm处测定吸光度,根据标准曲线计算其含量。
3.1.2. 体外溶出度测定
根据2015版《中国药典》第四部要求,参照溶出度与释放度测定(通则0931第二法),精确称取布洛芬65 mg于溶出杯中,以900 ml水为溶出介质,温度为37℃,转速为100 r/min。分别在5、10、15、30、45、60、120 min经0.45 µm微孔滤膜过滤后取样5 ml,同补加等体积的水 [5]。以水作为空白对照,在布洛芬最大吸收波长处测定吸光度,计算累计溶出百分率。
3.2. 布洛芬固体分散体的制备
3.2.1. 布洛芬物理混合物的制备
按最佳配方比例精密称定药物和载体,充分混合,精细研细后,过80目筛,备用。
3.2.2. 溶剂法
精密称定布洛芬原料药0.1000 g于25 ml的圆底烧瓶中,加入10 ml无水乙醇震荡至溶解完全,缓慢加入处方量的载体PVPP,磁力搅拌一定时间至混合均匀。一定温度下,水浴旋转蒸发混合溶液以除去大量无水乙醇,使其呈粘稠状态,所得产物于40℃烘箱干燥24 h,取出后研细,过80目筛,备用。
1) 考察载体用量
将布洛芬与PVPP (质量比1:1、1:3、1:5)的物理混合物置于圆底烧瓶中,磁力搅拌60 min,旋蒸温度40℃,制备固体分散体。所得产物的累积溶出度结果见图1。

Figure 1. Dissolution curves with different proportions of IBU by the solvent method
图1. 溶剂法中不同比例的IBU固体分散体溶出曲线图
载体用量对固体分散体的体外溶出有影响。在图1中,同一制备条件下,不同载体用量所制备SD的累积溶出度随载体比例的增加呈上升趋势,其中药物:载体 = 1:5时,2 h即可溶出87.60%,比原料药提高了2.48倍,且相比其他载体比例的SD溶出度的增幅较大。因此,确定布洛芬与PVPP的最佳质量比是1:5。
2) 考察旋转蒸发温度
将布洛芬与PVPP (质量比1:5)的物理混合物置于圆底烧瓶中,磁力搅拌60 min,旋蒸温度分别为40℃、50℃、60℃,制备固体分散体。所得产物的累积溶出结果见图2。
试验发现,旋转蒸发温度是影响固体分散体的体外溶出速率的一个因素。在图2中,在其他制备条件相同的条件下,随旋蒸温度升高,SD的累积溶出度有所降低,结果显示旋蒸温度为40℃制备的SD的效果较好,这可能是由于温度过高,溶剂还未充分分布在载体表面或内部空隙中就被除去,没有很好地提高药物的分散性所致。因此,确定布洛芬与PVPP的最佳旋蒸温度是40℃。
3) 考察磁力搅拌时间
将布洛芬与PVPP (质量比1:5)的物理混合物置于圆底烧瓶中,磁力搅拌时间分别设置为30 min、60 min、90 min,旋蒸温度40℃,制备固体分散体。所得产物的体外溶出结果见图3。
磁力搅拌时间也是固体分散体体外溶出的影响因素之一。在图3中,在其余制备条件相同的条件下,磁力搅拌时间90 min所制备的SD溶出效果最好,搅拌时间每延长30 min,可提高累积溶出度约7%。这是药物和载体充分接触,很好地分布在载体地表面及内部间隙的结果。因此,确定布洛芬与PVPP的最佳磁力搅拌时间是90 min。

Figure 2. Dissolution curves with different rotary evaporation temperatures of IBU by the solvent method
图2. 溶剂法中不同旋转蒸发温度的IBU固体分散体溶出曲线图

Figure 3. Dissolution curves with different magnetic stirring time of IBU by the solvent method
图3. 溶剂法中不同磁力搅拌时间的IBU固体分散体的溶出曲线图
3.2.3. 熔融法
精密称定布洛芬原料药0.1000 g和处方量的载体PEG6000于25 ml的烧杯中,均匀混合后得到物理混合物。再将烧杯放在水浴锅中,在制备的温度下搅拌一定时间后,迅速放在冰水中骤冷5 min。所得产物于40℃烘箱干燥24 h,取出后研细,过80目筛,备用。
1) 考察载体用量
将布洛芬与PEG6000 (质量比1:1、1:3、1:5)的物理混合物置于烧杯中,按“2.2.3项”制备固体分散体,熔融温度100℃,熔融时间30 min。所得固体分散体体外溶出结果见图4。
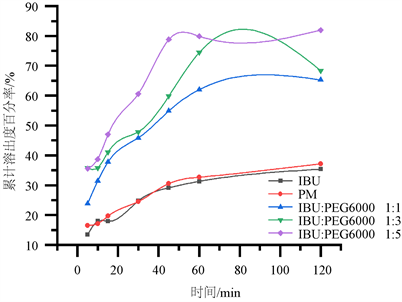
Figure 4. Dissolution curves with different proportions of IBU by the melt method
图4. 熔融法中不同比例的IBU固体分散体溶出曲线图
由图4结果可知,同一制备条件下制备不同载体比例的SD累积溶出度均高于IBU和相应的PM。这表明,随载体比例的增加,累计体外溶出度呈增长趋势,有利于SD的体外溶出。因此,确定布洛芬与PEG6000的最佳质量比是1:5。
2) 考察熔融时间
将布洛芬与PEG6000 (质量比1:5)的物理混合物置于烧杯中,按“2.2.3项”制备固体分散体,熔融温度100℃,熔融时间分别为15 min,30 min,45 min。所得固体分散体体外溶出结果见图5。
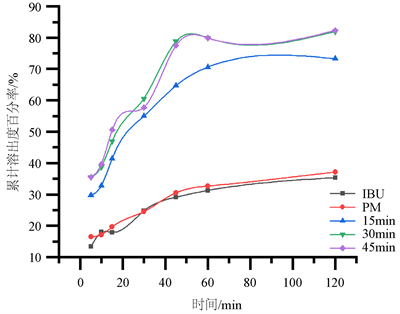
Figure 5. Dissolution curves with different melting time of IBU by the melt method
图5. 熔融法中不同熔融时间的IBU固体分散体溶出曲线图
由图5结果可知,在其他条件不变的情况下,熔融时间对固体分散体体外溶出有影响。随熔融时间的延长,产品的累积溶出度有所上升。在熔融时间为15 min时,SD的累积溶出度较低;在熔融时间为30 min和45 min时,SD的累积溶出度随熔融时间增长而上升,但增幅只有0.3%。因此,确定布洛芬与PEG6000的最佳熔融时长是30 min。
3) 考察熔融温度
将布洛芬与PEG6000 (1:5)的物理混合物置于烧杯中,按“2.2.3项”制备固体分散体,熔融温度分别为80℃,90℃,100℃,熔融时间30 min。所得固体分散体体外溶出结果见图6。
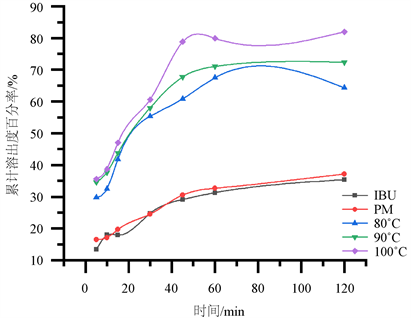
Figure 6. Dissolution curves with different melting temperatures of IBU by the melt method
图6. 熔融法中不同熔融温度的IBU固体分散体溶出曲线图
由图6结果可知,同一制备条件下,改变熔融温度,所得产品的累积溶出度存在差异。随熔融温度的上升,SD累积溶出度呈增加趋势且增幅较大,当熔融温度达到100℃时,SD的累积溶出度可达81.94%,最大增幅值为17.62%。因此,确定布洛芬与PEG6000的最佳熔融时间为100℃。
3.3. 布洛芬固体分散体的物相鉴别
3.3.1. 差示热扫描分析DSC
图7是布洛芬、布洛芬-PVPP物理混合物和固体分散体的DSC图谱。
布洛芬在77.01℃处存在尖锐的熔化吸热峰;PVPP与布洛芬的物理混合物(PM)在该位置也出现了一
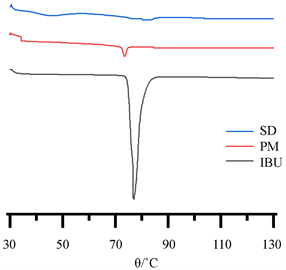
Figure 7. DSC pattern of IBU, IBU-PVPP physical mixtures and solid dispersions
图7. 布洛芬、物理混合物和固体分散体的DSC图谱
个较小的吸热峰,说明物理混合可在一定程度上减少药物结晶的产生,但大部分药物仍以原有晶型存在;而在固体分散体曲线中,布洛芬的吸收峰消失,由此可推出,SD中的原料药已转化为无定形或分子形态。
3.3.2. 粉末X射线衍射分析XRD
图8是布洛芬、布洛芬-PVPP物理混合物和固体分散体的XRD图谱。
布洛芬在6.05,11.74,15.88,16.86,18.59,19.28,21.29˚等位置出现高清晰的狭窄衍射峰,说明IBU以晶体形式存在;物理混合物中布洛芬的特征峰有不同程度的减弱和偏移,说明有部分的布洛芬以非晶体形式存在;在固体分散体中无明显的布洛芬晶体特征衍射峰,说明药物失去了原来的晶型,以无定形或分子形态存在于载体中。
3.3.3. 扫描电子显微镜分析SEM
图9表明,布洛芬大多以规则的棒状结晶存在且形态清晰可见;图10表明,布洛芬-PVPP物理混合物中,仍可见少量游离的药物晶体存在;图11表明,在以PVPP为载体的SD中已无IBU结晶存在,说明药物可能以无定形或分子形态高度分散在固体分散体中,这可能是由于IBU与PVP之间存在的作用力破坏了药物的晶型,也可能是制备SD时,有机溶剂(无水乙醇)在旋蒸过程中被除去,IBU立即被载体包合,抑制了其晶体的生长 [3]。
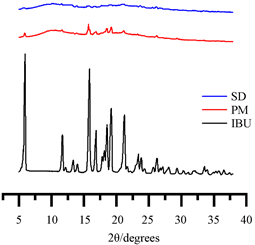
Figure 8. XRD pattern of IBU, IBU-PVPP physical mixtures and solid dispersions
图8. 布洛芬、物理混合物和固体分散体的XRD图谱

Figure 10. SEM results of IBU-PVPP physical mixtures
图10. 布洛芬-PVPP物理混合物的SEM结果

Figure 11. SEM results of IBU-PVPP solid dispersions
图11. 布洛芬-PVPP固体分散体的SEM结果
4. 结语
本文采用溶剂法和熔融法制备布洛芬固体分散体。溶出结果显示,以溶剂法为佳,载体用量增加、合适的旋蒸温度以及制备时间的延长都有利于固体分散体的体外溶出。这可能是由于布洛芬完全溶解在乙醇溶剂中,且乙醇充分地润湿了载体表面及其空隙,旋蒸除去溶剂后,药物以无定形形式附着在载体内部空隙和表面,提高了药物的表面积利用率,更好地改善了布洛芬的体外溶出效果 [6]。
通过对布洛芬固体分散体的DSC、XRD和SEM分析,可明显看出,布洛芬原料药主要以晶体的形式存在,分子间紧密堆积,粒子间的结合强度大,表面自由能较小,溶剂化作用小,故导致溶出效果不好。将其制备成固体分散体,可降低分子之间的堆积密度,增大药物的接触面积,达到改变晶型的目的,以提高药物的溶出速率。
尽管固体分散体技术是解决难溶性药物生物利用度低、溶解度差等难题的有效手段之一,但是SD的制备仍面临处方设计不明确、工艺不成熟、物理稳定性差等限制产品商业化的问题,其中物理稳定性差为最主要的因素 [7]。许多固体分散体经长期贮存后会出现硬度黏度增加、析晶或转晶等老化现象,从而导致药物溶出度降低 [8]。未来SD的研究重点应该更多放在SD稳定性机制上,从载体的选择、处方设计、制备方法等方面改善药物和载体相互聚集发生迁移的老化问题,从而更好地提高生物利用度,为SD的商业化提供可靠的依据。
基金项目
国家自然科学基金(NFSC 21562014);贵州大学SRT (No.191)计划。
NOTES
*通讯作者。