1. 引言
超分子化学是研究两种以上的化学物种通过分子间相互作用缔结而成的具有特定结构和功能的分子集聚体的科学。分子间的非共价相互作用又被称为弱相互作用,在理论和实验方面都是超分子化学研究的核心领域。当前对于超分子体系非共价相互作用的理论和应用研究主要集中在两个方面:氢键和σ-hole相互作用 [1];π相互作用 [2]。氢键和σ-hole相互作用可采用通式D···X-Y来表达。D为电子供体,可以是含有孤对电子或π电子的中性分子,也可以是带有负电荷的阴离子。相较于X,Y为具有较强吸电子能力的原子或基团。当X为氢原子时,D···H-Y即为大家所熟知的氢键。氢键是最早实际广泛运用于超分子体系的分子间非共价相互作用。除了作为氢原子外,X还可以是第IV、V、VI、VII主族元素的原子,统称为σ-hole相互作用。
当X为第VII主族元素的原子时,此分子间作用被称为卤键。类似于氢键,卤键也已经作为超分子体系中一种常见的非共价相互作用,广泛运用于晶体工程 [3] 和药物设计各领域 [4]。当X为第VI主族元素的原子时,此分子间作用被称为硫键 [5]。当X为第V主族元素的原子时,此分子间作用被称为磷键 [6] [7] [8] [9]。当X为第IV主族元素的原子时,此分子间作用被称为硅键(tetrel bond) [10]。尽管硫键、磷键、硅键存在于分子晶体中的现象发现得较早,但是早期研究者没有对这些非共价相互作用进行命名,而且也没有进一步研究这些作用力的本质及其在超分子体系中的应用可能。理论化学以及超分子化学研究群体对这些被重新“发现”的相互作用进行广泛和深入的研究是最近十年的事 [1]。
近两年的硅键理论计算研究已有较多发现,这些研究包括各种电子供体:含孤对电子的分子 [11]、阴离子 [12]、烯烃或炔烃 [13]、金属氢化物 [14]、自由基 [15]。但据我们所知,当前对于芳香环化合物作为电子供体的硅键理论计算研究还未见报道。前面的研究表明芳香环化合物作为电子供体可以有效参与形成卤键 [16] 和磷键 [17],那芳香环化合物作为电子供体可以有效参与形成硅键吗?本文通过量子化学计算方法对H3XF (X = Ge, Si)和五种芳香环化合物(苯、吡啶、吡咯、呋喃、噻吩)可能形成的硅键体系进行研究,以期进一步加深和扩展对硅键的认识。超分子化学是研究两种以上的化学物种通过分子间相互作用缔结而成的具有特定结构和功能的分子集聚体的科学。分子间的非共价相互作用又被称为弱相互作用,在理论和实验方面都是超分子化学研究的核心领域。当前对于超分子体系非共价相互作用的理论和应用研究主要集中在两个方面:氢键和σ-hole相互作用 [1];π相互作用 [2]。氢键和σ-hole相互作用可采用通式D···X-Y来表达。D为电子供体,可以是含有孤对电子或π电子的中性分子,也可以是带有负电荷的阴离子。相较于X,Y为具有较强吸电子能力的原子或基团。当X为氢原子时,D···H-Y即为大家所熟知的氢键。氢键是最早实际广泛运用于超分子体系的分子间非共价相互作用。除了作为氢原子外,X还可以是第IV、V、VI、VII主族元素的原子,统称为σ-hole相互作用。
2. 计算方法
采用MP2方法和aug-cc-pVDZ基组进行结构优化和频率分析,用aug-cc-pVTZ基组对优化结构做单点能计算,相互作用能用Counterpoise方法 [18] 做了基组重叠误差(BSSE)矫正。所有上述计算和自然键轨道(NBO)分析 [19] 用Gaussian 09软件 [20] 完成。静电势图采用GaussView绘制 [21],静电势的极大和极小值计算、分子中的原子(AIM)分析 [22] [23]、约化密度梯度函数(RDG)分析 [24] 使用Multiwfn软件 [25] 完成,RDG填色等值面图采用VMD程序 [26] 进行绘制。
3. 结果与讨论
3.1. 静电势
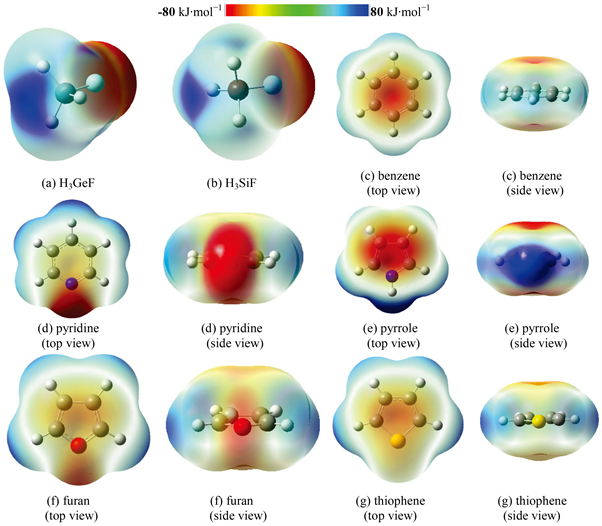
Figure 1. Electrostatic potential surfaces of H3XF and aromatic ring compounds
图1. H3XF和芳香环化合物的静电势
图1显示了H3GeF、H3SiF和芳香环化合物的分子表面静电势图。图1(a)和图1(b)分别为H3GeF和H3SiF的静电势图,由图可看出沿着F-Ge和F-Si σ键延长线,在Ge和Si原子附近存在呈蓝色的正静电势区域(σ-hole)。仔细观察还可发现,H3GeF的σ-hole区域比H3SiF的σ-hole区域大一些,且颜色深一些。进一步的计算表明H3GeF和H3SiF的σ-hole区域最大正静电势值分别为190和173 kJ∙mol−1。
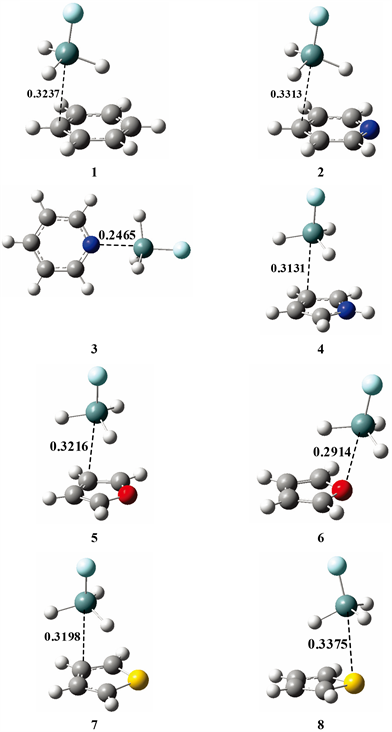
Figure 2. Structures of complexes 1-8
图2. 复合物1-8的结构
五种芳香环化合物的静电势图大体上可分为两类。第一类为苯、吡咯、噻吩,分别如图1(c)、图1(e)、图1(g)所示。这一类的特点是在芳香环上、下方对称分布着一片呈红色的负静电势区域(来源于芳香环离域π电子),且颜色深度为吡咯 > 苯 > 噻吩,三者的最小负静电势值分别为−98、−67、−64 kJ∙mol−1。第二类为吡啶和呋喃,这一类的特点是除了在芳香环上、下方存在负静电势区域外,在杂原子(N和O)附近的环侧面方向还存在另一负静电势区域(来源于芳香环杂原子未参与成键的孤对电子)。吡啶的静电势如图1(d)所示,环侧面(深红色区域)和环上、下方(浅黄色区域)的最小负静电势值相差很大:分别为−152和−32 kJ∙mol−1。呋喃的静电势如图1(f)所示,环侧面(浅红色区域)和环上、下方(微红色区域)的最小负静电势值则较为接近:分别为−77和−60 kJ∙mol−1。

Table 1. Structural parameters and interaction energies of complexes
表1. 复合物的结构参数和相互作用能
3.2. 结构及其相互作用能
图2显示了H3GeF与芳香环化合物形成的8个硅键复合物结构(1-8),这些复合物结构的一些重要结构参数和相互作用能列在表1中。H3SiF与芳香环化合物形成的复合物(9-16)的结构参数和相互作用能也列在表1中,仔细观察可以发现H3GeF的8个硅键复合物的相互作用能比相应的H3SiF复合物的作用能要大一些(例如结构1和9的作用能分别为−17.39和−15.50 kJ∙mol−1),这与我们前面提到的图1(a)和图1(b)的静电势分布相一致(H3GeF的σ-hole区域最大正静电势值要比H3SiF大一些)。由于H3SiF复合物的结构特征和相互作用能变化规律与相应的H3GeF复合物极其相似,因此下面主要以H3GeF复合物为例进行分析。
为了便于讨论,先对表1中的结构参数进行说明。RX-Y为复合物的平衡距离,X代表H3GeF、H3SiF分子中的Ge或Si,Y则为芳香环化合物中与X距离最近的原子,图2用虚线标出了8个H3GeF硅键复合物的RX-Y。按照Y原子的种类,可以将复合物的结合模式分为两类:若Y为碳原子,则命名为结合模式I;若Y为杂原子(N、O、S),则命名为结合模式II。按照结合模式I形成的复合物中,Ge或Si主要与碳原子发生硅键相互作用,模式II则主要与杂原子发生硅键相互作用。∠FXY体现了复合物中硅键作用的方向性。ΔRF-X为H3GeF、H3SiF与芳香环化合物形成硅键复合物前后,F-Ge或F-Si的键长改变值。下面我们首先概述8个H3GeF硅键复合物结构,然后再结合静电势对复合物结构和相互作用能进行分析。

Figure 3. Correlation between the interaction energies of H3GeF complexes (mode I) and the electrostatic potential of corresponding aromatic ring compounds
图3. H3GeF的模式I复合物的结合能与对应的芳香环化合物静电势的相关图
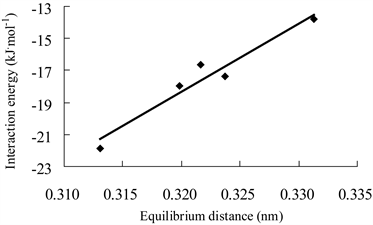
Figure 4. Correlation between the interaction energies of H3GeF complexes (mode I) and equilibrium distances
图4. H3GeF的模式I复合物的结合能与平衡距离的相关图
由于苯只有碳原子而没有杂原子,所以只能按照模式I与H3GeF形成硅键复合物(图2中的1)。与苯不同,吡啶可以形成两种模式的复合物(图2中的2和3),模式II的复合物(3)中,H3GeF处于环的侧面。尽管吡咯与吡啶都是含氮原子的芳香杂环化合物,但吡咯只能形成模式I复合物(图2中的4)。与吡啶的状况类似,呋喃也可形成两种模式的复合物(图2中的5和6),但在模式II的复合物(6)中,H3GeF处于环上方偏外的方向而不是环的侧面。噻吩也可形成两种模式的复合物(图2中的7和8),在模式II的复合物(8)中,H3GeF处于环上方偏内的方向。
上述结构大多数可以通过芳香环静电势进行合理解释,但是也有一些结构得不到明晰解释:1) 如同下面NBO分析一节讨论中所指出的那样,结合模式I的电子供体为碳碳π键,碳碳π键参与构成芳香环离域大π键,而离域大π键是芳香环上、下方的负静电势区域产生的根源,图1显示五种芳香环上、下方都存在负静电势区域,与五种芳香环都能形成模式I复合物相一致。2) 如图1(e)所示,吡咯负静电势区域主要集中于碳原子而不是氮原子,所以吡咯只能形成模式I复合物。3) 吡啶在芳香环上、下方和环侧面方向各存在一负静电势区域,故吡啶在这两个方向上分别形成了两种模式的复合物。4) 与吡啶不同,呋喃的模式II复合物中,H3GeF不在环侧面而在环上方偏外方向,这可能与呋喃两个负静电势区域的最小值差别不大有关(吡啶则有显著差异),H3GeF在呋喃模式II复合物中的取向似乎是呋喃两个负静电势区域综合影响的结果。5) 噻吩虽然只有一个负静电势区域,但也存在两种复合物。从上面的分析可以看到,静电势虽然对于理解复合物结合模式有很大帮助,但是单从静电势出发还不能精准预测所有复合物的结构特征。静电势不但能解释复合物结合模式,静电势的数值还与复合物的相互作用能紧密相关。图3显示了5个H3GeF模式I复合物的结合能与对应的芳香环负静电势区域(环上、下方区域)的最小负静电势值之间呈现明显相关,线性相关系数R2 = 0.979。
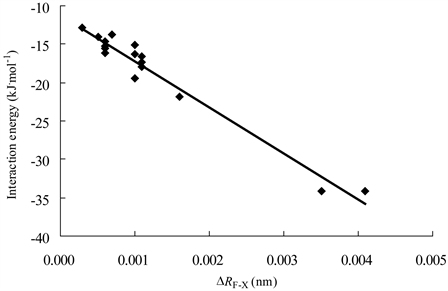
Figure 5. Correlation between the interaction energies of complexes and ΔRF-X
图5. 复合物的结合能与ΔRF-X的相关图
由表1所列各复合物相互作用能可看出一些重要结果。H3GeF和H3SiF与五种芳香环化合物形成模式I复合物,稳定性顺序为:吡咯 > 噻吩 > 苯 > 呋喃 > 吡啶。H3GeF和H3SiF只与三种芳香环化合物形成模式II复合物,稳定性顺序为:吡啶 > 噻吩 > 呋喃。呋喃和噻吩的两种复合物稳定性顺序为模式I > 模式II,而吡啶的两种复合物稳定性顺序为模式II > 模式I。在所有的复合物中,吡啶模式II复合物稳定性最强而吡啶模式I复合物稳定性最弱。
16个复合物的平衡距离在0.23至0.34 nm之间,其中两个吡啶模式II复合物(3和11)的平衡距离明显小于其它复合物,而它们的结合能明显大于其它复合物。图4显示了5个H3GeF模式I复合物的结合能与对应的平衡距离之间呈现线性相关,R2 = 0.937。所有16个复合物的∠FXY均大于172˚,表明硅键作用具有接近直线的方向性。复合物的ΔRF-X均为正值,表明H3GeF、H3SiF形成硅键复合物后,F-Ge或F-Si都增长,其中两个吡啶模式II复合物增加值最为显著。图5显示了16个复合物的结合能与对应的ΔRF-X值之间呈现较高线性相关,R2 = 0.962。
3.3. NBO、AIM和RDG分析
表2列出了8个H3GeF复合物的NBO分析结果:复合物的主要供–受体轨道及其对应的二阶稳定化能(E(2))、Wiberg键级(WBI)和电荷转移量(qCT)。需要说明的是复合物6的稳定化能为两组供–受体轨道作用值之和,对于这点我们还会进行详细讨论。所有8个复合物的受体轨道均为σ*(F-Ge),形成复合物意味着σ*(F-Ge)反键轨道会有部分占据,σ(F-Ge)成键轨道削弱,这与我们前面提到复合物中F-Ge都增长相一致。
从表中还可以看出所有模式I复合物的供体轨道均为C=C π轨道,而所有模式II复合物的供体轨道均为孤对电子,因此按照NBO分析结果,可笼统的将模式I称为π型作用,而将模式II称为n型作用。但是NBO分析是一种定域化的分析,对于我们本文所涉及到的离域体系还需要结合具体复合物结构进行分析才能得到准确结果。将复合物3划为n型作用显然是准确的,因为参与构成复合物的电子是侧面氮原子孤对电子,而参与形成离域大π键的那对电子没有参与构成复合物。但是将复合物8划为n型作用却明显与事实不符,复合物8中参与构成复合物的电子是硫原子参与形成离域大π键的那对电子,而侧面的硫原子孤对电子没有参与构成复合物。对于类似于8这样的复合物结构,实际文献报道也的确有分歧,有人将其划为π型 [16] 作用的也有人将其划为n型 [17] 作用。我们认为还是根据实际结构特征将其划为π型作用为宜。将噻吩的两种复合物(7和8)都划分为π型作用,也与噻吩的静电势分布一致:只有源于芳香环离域π电子的负静电势区域(环上、下方),而没有源于未参与成键孤对电子的负静电势区域(环侧面)。
Table 2. NBO analysis results of H3GeF complexes
表2. H3GeF复合物的NBO分析结果
呋喃模式II复合物6的情况更为复杂一些,NBO分析表明结构6中,呋喃环氧原子的两对孤对电子对复合物的形成有大致相当的贡献:两对孤对电子与σ*(F-Ge)作用的二阶稳定化能分别为8.0和8.1 kJ∙mol−1。而结构3和8中氮原子和硫原子只有一对孤对电子对复合物的形成有显著贡献。也就是说,与吡啶和噻吩不同,呋喃参与形成离域π键和未参与成键的孤对电子都对复合物的形成有相当的贡献。因此我们建议将结构6划分为n/π混合型。将复合结构6归为n/π型也与我们前面根据静电势进行分析得到的结果一致:呋喃两个负静电势区域的最小值差别不大,H3GeF在呋喃模式II复合物中的取向可能是呋喃两个负静电势区域综合影响的结果。此外,对比图1中复合物结构3、6、8的H3GeF取向(环侧面、环上方偏外、环上方偏内)也的确能看出从n型到n/π型再到π型的过渡。
Table 3. AIM analysis results of H3GeF complexes
表3. H3GeF复合物的AIM分析结果
复合结构3的电子密度(ρ)和电子密度的拉普拉斯量(
ρ)均比其它结构明显大很多,这与它的相互作用能明显更大相一致。相互作用能与E(2)、WBI和qCT都有一定相关性,线性相关系数R2分别为0.906、0.861和0.901。如图6、图7、图8所示。

Figure 6. Correlation between the interaction energies of complexes and E(2)
图6. 复合物的结合能与E(2)的相关图
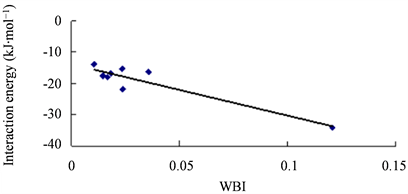
Figure 7. Correlation between the interaction energies of complexes and WBI
图7. 复合物的结合能与WBI的相关图
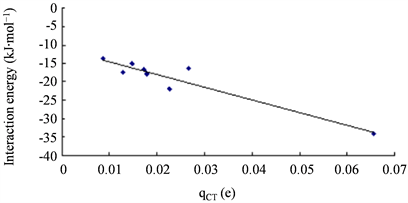
Figure 8. Correlation between the interaction energies of complexes and qCT
图8. 复合物的结合能与qCT的相关图
表3列出了8个H3GeF复合物的AIM分析结果:复合物键临界点的电子密度(ρ)、电子密度的拉普拉斯量(
ρ)和总能量密度(H)。复合物的
值在0.007至0.035之间,符合弱相互作用的特征。
值常用于表征相互作用的强度,对比表1和表3,可以看到相互作用能最大(3)和最小(2)的两个复合物,其
值也分别为最大和最小。8个复合物的相互作用能与
值有一定相关性,线性相关系数R2为0.889。复合物的
ρ值为正,表明复合物的硅键相互作用属于闭壳层相互作用。8个复合物中只有结构3的H值为负,这意味着结构3的硅键相互作用具有一定共价特征,这与结构3具有较大的相互作用能和较短的平衡距离一致。
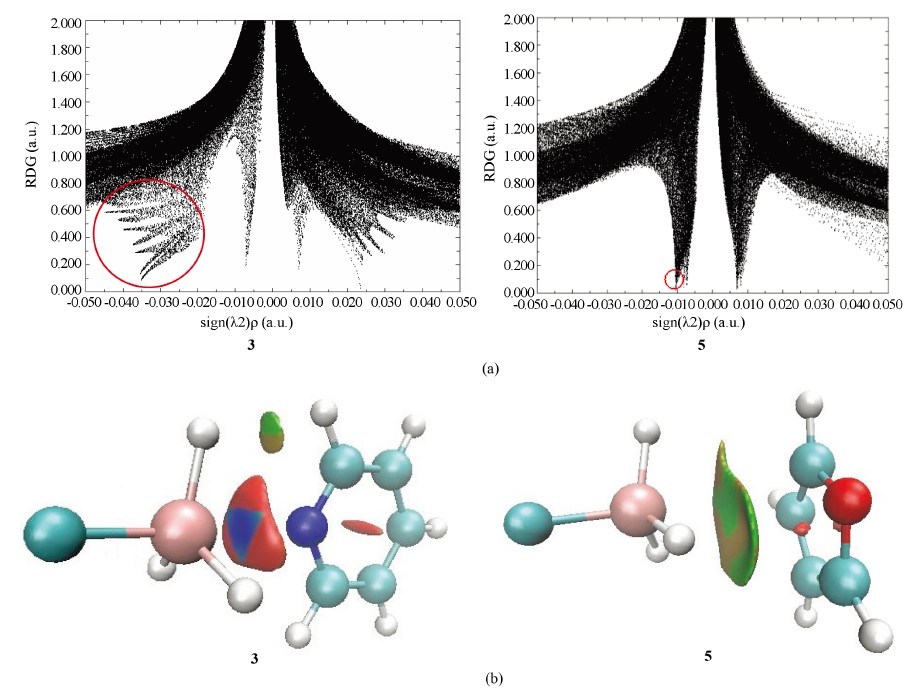
Figure 9. The scatter diagram (a) and color-filled isosurface (b) of RDG analysis for complexes 3 and 5
图9. 复合物3和5的RDG分析散点图(a)和填色等值面图(b)
RDG分析能形象显示非共价相互作用,这里我们以复合物结合能较大的3和复合物结合能较小的5为例进行说明,图9显示了复合物3和5的RDG散点图和填色等值面图。RDG散点图中,左侧(sign(λ2)ρ < 0)突起部分代表非共价相互作用,且数值越小,相互作用越强,右侧(sign(λ2)ρ > 0)突起代表排斥作用,中间突起代表范德华相互作用。图中圈出了代表复合物3和5的硅键相互作用的近似区域,复合物3在sign(λ2)ρ为−0.02至−0.04 a.u.之间有明显突起,而复合物5在sign(λ2)ρ为−0.01 a.u.处有突起,表明复合物3的硅键相互作用强于5,与它们的相互作用能数值相符。RDG填色等值面图主要有蓝、绿、红三色,蓝色意味着较强吸引作用,绿色意味着较弱吸引作用,红色意味着较强排斥作用。结构3在锗原子和氮原子之间存在蓝色区域,表明此结构中锗原子和氮原子之间有较强的硅键相互作用,结构5在锗原子和碳原子之间主要为绿色区域,表明此结构中锗原子和碳原子之间有较弱的硅键相互作用。注意复合物3在蓝色区域外还存在大片红色区域,意味着有较强排斥作用的存在,这显然是因为在复合物3中H3GeF与吡啶分子距离较近的缘故。这些红色区域在复合物3的散点图中,则对应着右侧区域那些代表排斥作用的大面积突起。
4. 结论
本文通过量子化学计算方法对H3GeF和H3SiF与五种芳香环化合物形成的硅键体系进行了研究。静电势分析表明H3GeF和H3SiF具有被称为“σ-hole”的正静电势区域,而五种芳香环化合物则各具有一个或两个负静电势区域。H3SiF复合物的结构特征和相互作用能变化规律与相应的H3GeF复合物相似,但H3GeF复合物的相互作用能比相应的H3SiF复合物略大。复合物的结合模式可分为两类,模式I复合物中Ge或Si主要与碳原子发生硅键相互作用,模式II则主要与杂原子发生硅键相互作用。H3GeF和H3SiF与五种芳香环化合物都能形成模式I复合物,稳定性顺序为:吡咯 > 噻吩 > 苯 > 呋喃 > 吡啶。H3GeF和H3SiF只可与三种芳香环化合物形成模式II复合物,稳定性顺序为:吡啶 > 噻吩 > 呋喃。呋喃和噻吩的两种复合物稳定性顺序为模式I > 模式II,而吡啶的两种复合物稳定性顺序为模式II > 模式I。在所有的复合物中,吡啶模式II复合物稳定性最强而吡啶模式I复合物稳定性最弱。通过具体考察复合物结构并结合NBO分析,发现所有模式I复合物均为π型作用,而模式II复合物则各不同:吡啶模式II复合物为n型作用,呋喃模式II复合物为n/π型作用,噻吩模式II复合物为π型作用。AIM分析表明吡啶模式II复合物的硅键相互作用具有部分共价特征而其它复合物则无共价特征。RDG分析形象化的展示了吡啶模式II复合物和呋喃模式I复合物分别存在较强和较弱的硅键相互作用。
基金项目
云南省大学生创新创业训练计划项目(2077360152),云南省教育厅科学基金资助项目(2015Y434)。
NOTES
*通讯作者。