1. 引言
在现代科学研究中,分子模拟技术已经变得越来越重要,它的发展很大程度上为科学家们提供了便利。而对于绝大部分有机反应而言,催化剂是决定其反应速率的主要因素,作为部分催化剂的活性成分,过渡金属得到了众多科学家们的关注,然而基本的催化机制与反应机理仍然不能通过简单的实验得到。分子模拟技术在分子筛催化剂、固体化学、无机材料研究开发领域的应用已非常广泛,大量的文献报道了利用分子模拟技术建立催化剂的结构模型,研究催化剂的吸附、催化机理,以及推测新型催化剂结构方面的研究成果。使用分子模拟方法我们可以得到与实验现象相吻合的理论数据,通过进一步的模拟计算达到预测实验现象继而指导实验思路的目的。
2. 分子模拟方法
近年来,随着高技术学科的飞速进步,各项科学研究都发生了翻天覆地的变化。化工学科也不例外,除多年来已形成的理论和实验研究之外,一种完全独立而新颖的研究手段——分子模拟飞速发展起来。纵观过去的20年,随着计算机硬件和算法的发展,分子模拟在化学,制药,材料等相关的工业中发挥着越来越重要的作用。
目前,科学家和工程师们对分子模拟抱有很高的期望,在美国化学会、化工学会、化学品生产协会等发布的2020年技术展望中,它被认为是到2020年实现化学工业从产品到过程设计完全自动化的一个关键技术。分子模拟之所以受到这样的重视,与它自身的特点和相关学科的发展是密不可分的。
随着分子模拟方法和高性能计算的高速发展,分子模拟已经可以运用于很多重要的问题研究中。这种优势在于:在进行昂贵的实验合成,表征,加工,组装和测试之前先利用计算机进行材料的设计,表征和优化,理论和模拟可以预测出目前实验条件所无法测出的结果,并对整个新材料的合成、设计进行高效周全的思考。
密度泛函理论
当使用分子模拟方法用来计算分子和固体的电子结构和总能时存在两种不同的方法:基于波函数的方法和密度泛函理论方法。前者如果有高水平的构型相互作用,就能够得到非常精确的结果,当前计算规模上限为10~100个电子。当考虑过渡金属表面时,因为每个过渡金属有数量级为10的价电子数,这表明能够处理的原子数在10左右。这使得该方法在需要用复杂模型模拟的催化剂常规处理方面没有吸引力。存在在低精度外围区内嵌入高精度区域的精细方法,来提高体系尺寸,让波函数方法可用。这类计算存在成本和体系尺寸限制说明,在表面科学和催化反应中,它是基本方法,用来作为标准,用于校准低计算量DFT方法的精度。所以在这我们只讨论DFT方法。
密度泛函理论全部建立在由Kohn和Hohenberg所证明的两个基本数学定理,以及由Kohn和Sham在1960年代中期所推演的一套方程的基础上。两个基本定理分别为:从Schrödinger方程得到的基态能量是电荷密度的唯一函数;基态电荷密度唯一决定了基态的所有性质,包括能量和波函数。
DFT方法描述如何通过解一组单电子薛定谔方程(K-S方程)来获得基态电荷密度和总能,而避免解更复杂的多电子薛定谔方程。这导致计算的极大简化,从而能够处理1000个电子以上的体系。
解K-S方程的方法有很多种。简单的说,他们可以通过描述表面的模型差异,所用基组的种类和交换关联影响的近似处理方法三种标准来做区分。
3. 过渡金属催化剂的应用
IUPAC是这样定义过渡金属的:原子的d带只有部分被填满的元素,或者可以用不完全的d亚外层产生阳离子的元素。由于拥有特殊的电子结构,不管是以元素或是离子形式存在,过渡金属往往具有不错的催化活性,因此过渡金属被广泛应用于化工领域。
3.1. 过渡金属在均相体系中的应用
在过去几十年里,过渡金属催化的均相有机合成反应受到了化学家的广泛关注并引起人们的极大兴趣,它改变了传统的有机合成方法,大大缩短了合成路径,因而过渡金属催化的均相有机合成反应一直占据着有机合成领域的主导地位 [1] 。
芳基之间键的形成反应是有机合成中最重要的工具之一,这个反应至今已有百余年的历史,并且是最早使用过渡金属作为催化剂的反应。这个领域的发展不仅仅是有机化学一个很好的例证,更是揭露了过渡金属催化剂的重要性 [2] 。
2001年10月10日,瑞典皇家学院将2001年的诺贝尔化学奖颁给不对称催化合成的3位先驱科学家 [3] 。其中来自威廉·诺尔斯的贡献是,他发现可以使用过渡金属来对手性分子进行氢化反应,以获得具有所需镜像形态的最终产品。他的研究成果很快便转化成工业产品,如治疗帕金森氏症的药L-DOPA就是根据诺尔斯的研究成果制造出来的。而野依良治的贡献是进一步完善了用于氢化反应的手性催化剂的工艺。巴里·夏普莱斯的成就是开发出了用于氧化反应的手性催化剂。这些基本的研究在很大程度上扩大了不对称合成的范围并允许更有针对性的药品合成,如抗生素、抗炎药物和其他药物制备。
2010年10月6日,Pd催化的Heck反应被授予了2010年的诺贝尔化学奖 [4] ,在合成化学中,它做到了增长碳链,并在先进功能材料和生物制药等多个领域广泛应用,已发展成为支撑有机电子、药物开发、精细化工等现代工业文明的重要手段。钯原子在反应中作为催化剂就像“媒人”一样,把不同的碳原子吸引到自己身边,使碳原子之间的距离变得很近,从而容易结合,而钯原子本身不参与结合也不会被消耗掉,反应不需要把碳原子激活到很活跃的程度,副产物比较少,也很容易除去,从而使反应变得更加精确和高效。
芳香族杂环化合物因其展现出特殊的生物、药物、与材料功能而成为一种重要的有机分子。近几年,碳氢键的直接功能化成为最直接有效的芳香族杂环化合物歧化的方法。然而,直接烷基化是十分困难的,Peng Ren等 [5] 人发现了一种含铜化合物能有效催化苯并噁唑的直接烷基化。
但是均相催化体系存在一些制约其发展的瓶颈问题,如:过渡金属催化剂的价格较为昂贵、不能重复使用、产物分离困难以及产物中含有痕量过渡金属催化剂残留等问题。随着药物合成化学和有机化学等对洁净化合成的严格要求,可用于液相合成的固载化催化剂愈来愈受到人们的青睐,这使得合成化学家们把更多的兴趣转移到可以回收再利用并且容易分离的负载型催化剂上。
3.2. 过渡金属在非均相体系中的应用
开发负载型过渡金属催化剂是解决均相催化剂较难分离与回收以及过渡金属残留等问题的主要策略之一。载体需选用具有来源广泛,价廉易得,溶剂适用性广,具有较好的热稳定性和机械强度,易于纯化和回收等优点的无机、有机或杂化材料。另外催化剂载体的使用为反应提供了一种全新的反应环境,为进一步研究催化反应历程提供了很好的思路。近年来许多负载型金属催化剂由此被设计合成,并对很多反应体系起到了较好的催化效果,有些催化剂已经表现出了具有工业应用的潜在价值。成功的负载型催化剂在反应中可以催化量使反应进行完全,其效果与均相催化体系效果相当,而且反应结束后,通过简单分离除去溶剂后即可得到产物,并回收可循环使用的载体催化剂。
相较于传统的液相化学,将过渡金属固定在载体上这种方式有很多优点。在理想状况下,背负载的复合物可以通过简单地过滤从反应混合物中分离出来,他们不会污染产物,他们可以回收再利用,并且他们可以提高反应选择性。由于过渡金属价格昂贵,将其负载在载体上可以使其更容易分离、回收、再利用,不仅实现了经济上的优势并且还易于控制。
碳碳键偶联反应在有机化学中是一个基本反应,许多能在温和的反应条件下完成转变的催化过程与方法都毫无例外的备受瞩目。其中一种过渡金属催化剂是通过将过渡金属或者金属配合物吸附于载体表面上来实现,另外一种是通过化学键联的手段,先在载体表面上接枝具有特殊功能的基团,再经由配位方式将过渡金属载于功能基团上。
2012年,Li和Change等 [6] 首次报道了氧化铝固载纳米钯的芳基硼酸和芳基重氮盐的Suzuki偶联反应,反应不需要额外添加碱和配体,以甲醇为溶剂,室温下进行即可。
2013年,Liu等 [7] 报道了无配体参与的Pd/C催化的芳基溴化物和芳基硼酸的Suzuki偶联反应。在室温条件下,各种取代基的芳基溴化物和芳基硼酸都能高产率地在50%乙醇水溶液中顺利完成,催化剂可循环利用10次后,其催化活性未见显著降低。该方法可用于含氟液晶化合物的合成。
过渡金属催化剂的载体除了简单的无机材料外还可以使用有机载体,有机高分子聚合物负载过渡金属催化剂一般般是在聚合物的单体中有目的地镶嵌,能够充当配体的原子或官能团,或者在已成型的功能化聚合物上接枝修饰引入配体,再经由配位方式将过渡金属载于引入的配体上。
聚苯乙烯是常用的聚合物载体之一,早在1978 年,Teranishi等 [8] 曾报道了聚苯乙烯经过负载过渡金属钯而制得钯的配合物,并成功应用于催化Heck反应。后来,Zhang小组 [9] 也成功合成了接枝有邻菲洛林的聚苯乙烯的载体;1997年,Jang [10] 首次将聚合物负载钯催化剂应用于Suzuki偶联反应;2000年,El-Sayed等 [11] 报道了聚乙烯吡咯烷酮(PVP)稳定的纳米钯催化剂;2004年Hu等 [12] 用一锅法制备了聚N,N-二烷基碳二酰亚胺负载的纳米钯催化剂;2006年,Fan等 [13] 合成并报道了一种高效含磷树枝状聚合物稳定的钯纳米粒子及其催化Suzuki偶联反应;2009年,Razler等 [14] 报道了在无配体条件下,聚乙二醇2000 (PEG-2000)负载钯催化Suzuki偶联反应;同年,Guo课题组 [15] 报道了聚2-氨基硫酚(PATP)负载纳米金催化的Suzuki偶联反应;2010年,Wang课题组 [16] 报道了一种高效的聚苯乙烯负载钯催化剂的芳基溴或芳基碘与芳基铋合物的交叉偶联反应。
4. 密度泛函理论在过渡金属催化剂中的应用
全氧化或部分氧化反应在工业应用上是一类价值数十亿美金的催化反应。这类反应过程的催化剂一般包括过渡金属,例如:Rh,Pd,Pt,Au或Ag作为活性物质。这些工业催化剂是一个长达十年优化过程的产物,但是本质上还是一个“试错”的方法。除去对社会的明显重要性,我们必须承认合理的催化剂设计在现在并不现实,这是由于不能从原子级别来理解催化剂表面的催化过程。这种理解的缺失不仅是由于工业催化剂的复杂性,也是由于缺少可以在严厉环境条件下提供工业催化剂原子信息的实验技术。
密度泛函理论可以从原子规模研究催化剂的工作机理。如果这些计算加入了(微型)表面过程的动力学模拟就可以深入理解影响催化剂性能的一些因素。这使得预测各种不同催化材料的趋势成为可能。
4.1. 对吸附性能的研究
研究小分子与过渡金属表面的相互作用十分有意义。MinJi等 [17] 研究了O原子在Pd(111),Au(111)和Pd-Au(111)合金表面的化学吸附,用DFT-GGA方法找到了最合适的吸附位,用d带理论解释了成键强度和合金组成的关系;M.P.Andersson等 [18] 用DFT-D2方法比较了几种分子在Pt(111)面上的吸附能并与实验值进行了对比;Qiu M [19] 用第一性原理研究了CO2在一系列合金表面的化学吸附行为;D.Vasic等 [20] 研究了H2在过渡金属(111)面上的吸附和在Pt(111),Pt(100)及Pt(110)面覆盖率从0.25到1的吸附,发现H2的吸附能随着覆盖率的升高而降低。;Hongqing Shi和Catherine Stmpfl [21] 用DFT方法计算O在Au(111)面的吸附,研究包括在表面,内表面的吸附和表面氧化物的形成。
4.2. 对反应机理的研究
CO在Pd或Pt表面氧化是用来理解表面催化反应的基础步骤的标准之一。Gao等 [22] 用DFT理论研究了CO在Pt/γ-Al2O3上的氧化机理,在原子级别上研究了CO在Pt/γ-Al2O3催化剂上的氧化活性位及界面效应,为深入研究Pt/γ-Al2O3催化剂提供了有用信息;Hu Zhou等 [23] 研究了在不同CO覆盖度下CO在Pt团簇/TiO2(110)表面的氧化与吸附,为在现实或工业条件下理解CO在Pt团簇上氧化的机理提供了有用的信息;Hao等 [24] 通过实验和模拟手段研究了NO对CO在Pt,Pd,和PdAu等催化剂上的氧化的影响,用DFT理论计算了NO和CO在这些金属表面的吸附和反应性质,不仅揭示了电脑模拟的解释性与预测性,并且提供了催化剂表面的潜在催化机理。
4.3. 对反应活化能的研究
密度泛函理论也可以用来计算反应的活化能。Sippakorn Wannakao等 [25] 用DFT方法研究了C-H键在Au+与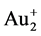 离子位于多种沸石的空位和内部时的断裂情况,发现具有最大孔径的工业催化剂Au-MCM-22有最高的活性(最低的活化能),因为它具有最稳定的过渡态;Natarajan Sathiyamoorthy等 [26] 用DFT方法比较了H2在不同过渡金属掺杂的Ni团簇上的吸附与脱附,发现掺杂Ph,Pd,Pt和Au可以降低反应活化能;Mercedes Boronat等 [27] 用DFT方法研究了用Au和Fe掺杂的锐钛矿TiO2,发现Fe掺杂的TiO2的氧活性更高。
离子位于多种沸石的空位和内部时的断裂情况,发现具有最大孔径的工业催化剂Au-MCM-22有最高的活性(最低的活化能),因为它具有最稳定的过渡态;Natarajan Sathiyamoorthy等 [26] 用DFT方法比较了H2在不同过渡金属掺杂的Ni团簇上的吸附与脱附,发现掺杂Ph,Pd,Pt和Au可以降低反应活化能;Mercedes Boronat等 [27] 用DFT方法研究了用Au和Fe掺杂的锐钛矿TiO2,发现Fe掺杂的TiO2的氧活性更高。
5. 结语
为解决均相催化反应中存在的产物不易分离、贵金属痕量残留以及催化剂的重复使用等问题,通过化学家们的不懈努力,制备了不同载体的负载型催化剂,在一定程度上解决了上述问题,为部分有机反应的工业化应用奠定了基础,同时使贵金属催化剂的工业化应用大大降低成本,也更有利于保护环境。但是这些负载催化剂仍有如下不足有待进一步解决:1) 在催化反应过程中,有金属的流失现象;2) 载体催化剂的活性和选择性需要提高;3) 发展负载有机小分子催化剂,用于催化不对称有机反应。
密度泛函理论为计算的精确度和费用提供了一个很好的平衡,并且成功应用于模拟一系列实验现象。然而,DFT理论和它未来的应用之中仍有许多挑战。这些挑战有科学上的也有技术上的,主要分为三个部分:1) 找到一个更通用的交换关联泛函,并将其用在寻找具体的化学反应机制;2) 解决不能适用于扩散作用(如范德华力);3) 将DFT理论应用于固态大体系。
基金项目
湖北省自然科学基金重点项目,(2013CFA091);武汉工程大学研究生教育创新基金,(CX2016097)。
NOTES
*通讯作者。