1. 引言
近年来,人们在超硬材料的研究方面取得了明显的进展。除人工合成了金刚石以外,还成功地合成了立方氮化硼,另外,作为新兴超硬材料的C3N4,近年来的研究也炙手可热。
众所周知,自然界存在的材料中金刚石的硬度最高。所以往往以金刚石设定硬度标准的上限,来衡量其他物质的硬度。然而随着科学研究的深入,这个概念已经从理论上受到严重的挑战。1989年,Liu和Cohen [1] 根据β-Si3N4的晶体结构,用C原子替换Si原子,在局域态密度近似下采用第一性赝势能带法从理论上预言了β-C3N4的硬度可以与金刚石媲美。这是人类第一次从理论上预言一种具有超硬性能的新材料。1996年,Teter等 [2] 通过计算认为C3N4可能具有5种结构,即α相、β相、立方相、准立方相以及类石墨相,并给出了它们的结构参数、体弹性模量、能量等参数。计算结果表明,这5种C3N4当中,除了类石墨相以外,其它4种相的硬度都接近或超过了金刚石硬度。
在理论的预言下,人们采用各种手段试图在实验室合成这种新型超硬材料。其合成方法现在已经有化学气相沉积法 [3] [4] [5] 、物理气相沉积法 [6] [7] 、高温高压合成法 [8] [9] 等。由于C3N4的化学惰性以及表征的困难,给其合成亦带来了极大的困难。如碳氮薄膜的制备大多情况下获得的是非晶态薄膜,难以获得单一晶相的薄膜;高温高压法中由于氮含量损失严重,通常难以获得理想的碳氮化学计量比;脉冲放电和高速冲击可以有效抑制氮含量的降低,但实验方法目前还处于探索阶段 [10] 。由于以上实验方法的各种缺陷,故对于C3N4性质的探索更倾向于计算模拟。
Duan等 [11] 采用基于密度泛函理论的LMTO-ASA能带计算方法对β-C3N4的能带结构进行了计算。马秋花等 [12] 采用第一性原理的密度泛函理论平面波赝势法,通过广义梯度近似对立方C3N4进行了研究,得出了立方C3N4具有较高的硬度、良好的化学稳定性、较强的红外线穿透性的结论。阮林伟等 [10] 采用第一性原理的密度泛函理论平面波赝势法,利用LDA和GGA泛函研究了α相、β相、立方相、准立方相以及类石墨相C3N4的能带结构及电学和光学性质。
从当前的研究现状来看,有不少学者对C3N4进行了理论研究,但大多集中于研究其能带结构、电学、光学、力学性质等,而对β-Si3N4转变为β-C3N4的过程中C原子替换Si原子的最可能路径以及两者弹性、硬度的对比等鲜有研究。因此,我们在β-Si3N4结构的基础上,用C原子不断替换Si原子,并寻找一个能量最低的替换路径,以得到最稳定的结构,基于密度泛函的第一性原理平面波超软赝势,运用广义梯度近似(GGA)势场进行计算,分析和比较了β-Si3N4和β-C3N4弹性、硬度的差异。
2. 计算参数与理论
2.1. 计算模型
本文所研究的β-C3N4的模型由C原子逐个替换β-Si3N4中的Si原子所得到。计算单元为一个原胞,每个原胞含有14个原子,其中6个原子为C原子或Si原子或它们的组合,8个原子为N原子。首先搭建β-Si3N4的模型,将模型中每个Si原子进行编号,如图1所示。任意选定第一个占位的Si原子(假设选1号Si原子),用C原子将其替换,优化结构,计算体系能量;然后用C原子替换余下5个Si原子中的一个,共有5种可能,分别对所产生的5种(C,Si)3N4进行结构优化,计算体系能量;选择体系能量最低的一种组合,再用C原子替换余下4个Si原子中的一个,此时有4种可能,对这4种结构进行优化,计算体系能量,选择能量最低的一种组合……以此类推,直到将所有的Si原子都替换成C原子,以此来确定替换过程中体系能量最低的路径。β-C3N4和 -Si3N4的结构相似,C原子和N原子均为SP3杂化,每个C原子和3个N原子相连,每个N原子和3个C原子相连。β-C3N4和β-Si3N4都为六方晶体,空间群为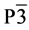 。
。
2.2. 计算公式
体系的总能量可以表示为对微小应变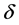 的泰勒级数展开形式
的泰勒级数展开形式
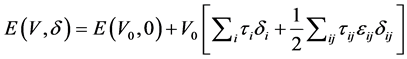 , (1)
, (1)
其中,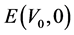 和
和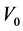 是未施加应变时体系的总能量和体积,
是未施加应变时体系的总能量和体积,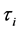 是应力张量元,而
是应力张量元,而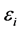 是Voigt因子。
是Voigt因子。
弹性常数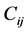 可表示为系统总能量对应变的二阶导数
可表示为系统总能量对应变的二阶导数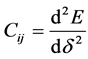 。
。
对于多晶系,Hill [13] 证明VRH (Voigt-Reuss-Hill)与实验结果更接近
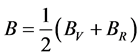 , (2)
, (2)
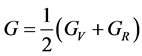 , (3)
, (3)
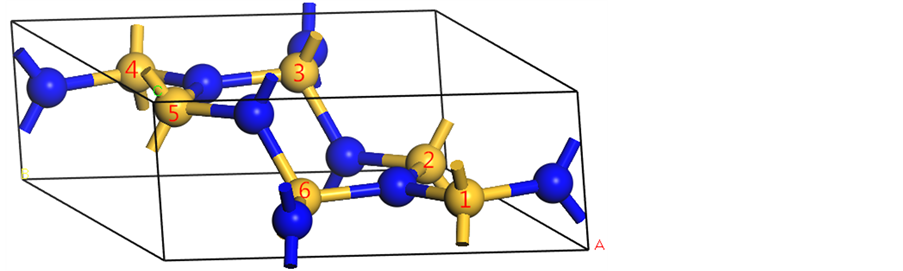
Figure 1. Structural diagram of β-Si3N4
图1. β-Si3N4的结构示意图( N,
N, Si)
Si)
式中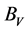 ,
,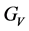 分别为Voigt模型体弹模量和剪切模量,
分别为Voigt模型体弹模量和剪切模量,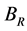 ,
,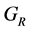 分别为Reuss模型体弹模量和剪切模量,可根据文献中相应的公式 [14] [15] 求得。
分别为Reuss模型体弹模量和剪切模量,可根据文献中相应的公式 [14] [15] 求得。
通过计算体弹模量和剪切模量的Hill值,我们进一步得到了多晶体系下各材料的杨氏模量和泊松比
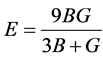 , (4)
, (4)
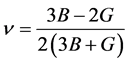 . (5)
. (5)
材料的德拜温度由下式计算求得 [16]
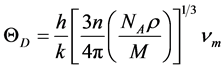 , (6)
, (6)
式中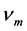 代表平均声学波波速,
代表平均声学波波速,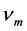 的公式 [17] 为
的公式 [17] 为
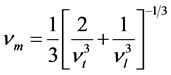 , (7)
, (7)
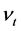 和
和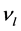 表示平均声学横波和声学纵波波速,其公式分别如下
表示平均声学横波和声学纵波波速,其公式分别如下
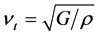 , (8)
, (8)
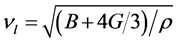 . (9)
. (9)
2.3. 计算参数
本文计算采用基于密度泛函理论(DFT) [18] 的第一性原理方法,由CASTEP [19] 程序完成。计算过程采用周期性边界条件,对交换关联势能采用了广义梯度近似(GGA)下的Perdew-Burke-Ernzerhof (PBE)方法 [20] ,晶体波函数由平面波基矢展开,并由超软赝势(Ultrasoft Pseudopotential, USPP) [21] 来实现离子实与价电子间的相互作用势。广义梯度近似(GGA)引入了电荷梯度来修正电荷密度的局域变化,并较大幅度地修正了少电荷密度区域的指数形式,在结构、总能、带隙等的计算中和实验结果符合较好。超软赝势(USPP)引入了广义的正交归一化条件,使得波函数变得更平滑,所需的平面波基底函数更少,仅需要非常小的截断能。
原子赝势计算考虑的外层电子组态为:C为2s22p2,N为2s22p3,Si为3s23p2。根据收敛性测试,我们选择波矢K空间中β-Si3N4和β-C3N4的平面波截断能780eV。根据收敛性测试,其布里渊区积分计算都为6 × 13 × 6的Monkhorst-Pack [22] 特殊K点对全布里渊区求和。当系统总能量变化稳定在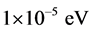 以内,并使得优化后作用在晶胞中每个原子上的力小于0.03 eV/Å,晶胞剩余应力低于0.05 GPa,公差偏移小于0.001 Å,认为达到收敛。结构优化计算所使用的晶格常数为实验值,运用BFGS算法 [23] [24] [25] [26] 先后对晶体模型结构及晶格原子的位置进行优化,以找出能量最低的结构,并在此基础上进一步计算其弹性和硬度。
以内,并使得优化后作用在晶胞中每个原子上的力小于0.03 eV/Å,晶胞剩余应力低于0.05 GPa,公差偏移小于0.001 Å,认为达到收敛。结构优化计算所使用的晶格常数为实验值,运用BFGS算法 [23] [24] [25] [26] 先后对晶体模型结构及晶格原子的位置进行优化,以找出能量最低的结构,并在此基础上进一步计算其弹性和硬度。
3. 计算结果与讨论
3.1. 能量最低路径的探索
为了找到 -Si3N4转变为β-C3N4的过程中C原子替换Si原子的最可能路径,即转变过程中能量最低的路径,本文先对晶胞进行结构优化,获得基态下的能量,再根据原子总数计算出单原子能,进一步比较替换过程中能量的高低。晶胞的体积优化采用非自旋极化方式。
经结构优化和计算,C原子替换Si原子过程中各种结构的单原子能量如图2所示。
根据图2,替换1号Si原子时,体系单原子能为−205.00 eV,替换第二个原子的5种可能中,替换4号Si原子时体系的单原子能最低,为−208.069 eV,替换第三个原子的4种可能中,替换2号Si原子时体系的单原子能最低,为−211.12040 eV,替换第四个原子的3种可能中,替换5号Si原子时体系的单原子能最低,为−214.285 eV,替换第五个原子的2种可能中,替换6号Si原子时体系的单原子能最低,为−217.38146 eV,最后替换仅剩的3号Si原子,此时体系单原子能为−220.643 eV。体系总能量的大小关系与单原子能大小关系相同,由此可见,当C原子按照图1中Si原子编号1→4→2→5→6→3的顺序依次进行替换时,体系在替换过程中的每一步都处于能量最低的状态。而能量最低时,体系在热力学上最稳定,此时替换最容易发生。故体系能量最低的路径就是β-Si3N4转变为β-C3N4的过程中C原子替换Si原子的最可能路径。
表1列出了结构转变过程中最可能替换路径(即路径1→4→2→5→6→3)的各结构的晶格参数、体积、密度以及键的个数。
从表1中可以看出,随着C原子替换Si原子的数目增多,晶胞体积逐渐减小。这主要是由于, C的原子半径小于Si的原子半径,C-N键的键长小于Si-N键的键长。随着C原子数目增多和Si原子数目减少,C-N键的数目增多而Si-N键的数目减少,因而晶胞体积呈减小趋势。
另外,在C原子取代Si原子的过程中,体系能量逐渐降低,表明体系在热力学上逐渐趋于稳定,β-C3N4的体系能量最低,因此最稳定。
综上,β-Si3N4转变为β- C3N4的过程中C原子替换Si原子的最可能路径为沿着图1中编号1→4→2→5→6→3依次进行替换。另外,随着C原子替换Si原子的数目增多,晶胞体积逐渐减小,体系能量逐渐降低,体系在热力学上逐渐趋于稳定。
3.2. 弹性对比
表2是β-C3N4和β-Si3N4的弹性刚度矩阵常数。前已述及,β-C3N4和β-Si3N4都是六方晶体。由广义胡克定律 [27] [28] [29] 可知,在弹性范围内,由于晶体结构本身的对称性,六方晶体的独立弹性常数为5个,分别为C11、C12、C13、C33和C44,其中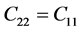 ,
,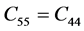 ,
,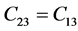 ,
,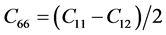 。根据六方结构晶体的弹性稳定性准则 [30]
。根据六方结构晶体的弹性稳定性准则 [30]
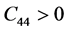 ,
, 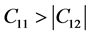 ,
, 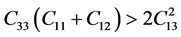 , (10)
, (10)
可以看出,β-C3N4和β-Si3N4的弹性常数值均能满足以上三个条件,说明β-C3N4和β-Si3N4的晶体结构都是稳定的。
一般而言,高硬度的材料具有较高的体弹模量,但剪切模量与材料的硬度更具关联性。表3列出了由GGA势场计算得到的β-C3N4和β-Si3N4的弹性模量E、体弹模量B、剪切模量G、泊松比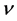 以及G/B和德拜温度
以及G/B和德拜温度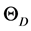 的值。体弹模量B、剪切模量G由VRH方法,即公式(2)和(3)计算得到。多晶体系下的杨氏模量E和泊松比
的值。体弹模量B、剪切模量G由VRH方法,即公式(2)和(3)计算得到。多晶体系下的杨氏模量E和泊松比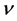 分别为(4)和(5)式的计算结果,
分别为(4)和(5)式的计算结果,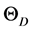 的值则由公式(6)~(9)计算得到。由表3可以看出,相比于β-Si3N4,β-C3N4的E、B和G都增大了一倍左右,这是由于C-N键的键长比Si-N键的键长更短,原子间结合力更强,随着C原子逐渐替代Si原子,C-N键的数目增多而Si-N键的数目减少,因而β-C3N4的弹性模量E、体弹模量B和剪切模量G都比β-Si3N4大。共价键材料,如金刚石,通常具有高剪切模量。一般地,共价键材料的泊松比大约为0.1,而金属材料的泊松比大约为0.33 [31] 。如果材料含有高度定向共价键,则剪切模量增加而泊松比会减小。
的值则由公式(6)~(9)计算得到。由表3可以看出,相比于β-Si3N4,β-C3N4的E、B和G都增大了一倍左右,这是由于C-N键的键长比Si-N键的键长更短,原子间结合力更强,随着C原子逐渐替代Si原子,C-N键的数目增多而Si-N键的数目减少,因而β-C3N4的弹性模量E、体弹模量B和剪切模量G都比β-Si3N4大。共价键材料,如金刚石,通常具有高剪切模量。一般地,共价键材料的泊松比大约为0.1,而金属材料的泊松比大约为0.33 [31] 。如果材料含有高度定向共价键,则剪切模量增加而泊松比会减小。
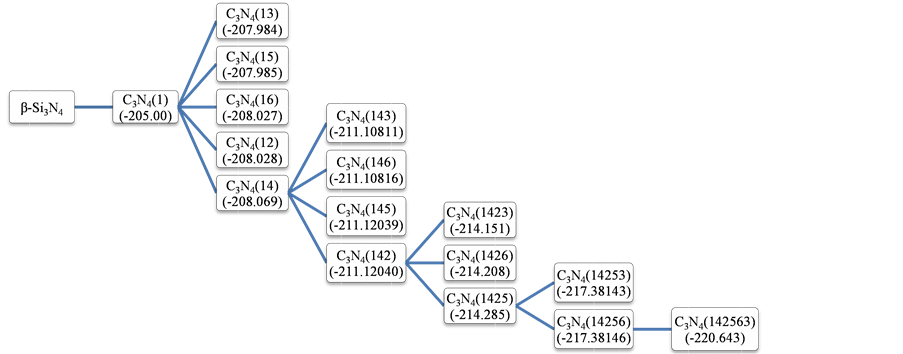
Figure 2. The single atom energy of various structures in the C atoms to replace the Si atom process
图2. C原子替换Si原子过程中各种结构的单原子能(eV)

Table 1. Lattice constants a, b, c (Å), α, β, γ (°),volume V (Å3), density ρ (g∙cm−3), System energy (eV) and bond number of β-C3N4, β-Si3N4 and (C,Si)3N4
表1. β-C3N4,β-Si3N4和(C,Si)3N4的晶格参数a,b,c (Å)、α,β,γ (°)、体积(Å3)、密度(g∙cm−3)、体系能量(eV)和键的个数
Table 2. Elastic stiffness constants (GPa) of β-C3N4 and β-Si3N4
表2. β-C3N4和β-Si3N4的弹性刚度矩阵常数(GPa)
Table 3. Elastic modulus E (GPa), bulk modulus B (GPa), shear modulus G (GPa), Poisson’s ratio, G/B and debye temperature (K) of β-C3N4 and β-Si3N4
表3. β-C3N4和β-Si3N4的弹性模量E (GPa)、体弹模量B (GPa)、剪切模量G (GPa)、泊松比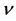 以及G/B和德拜温度
以及G/B和德拜温度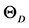 (K)
(K)
β-C3N4的泊松比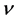 比β-Si3N4小,这说明β-C3N4的不可压缩性比β-Si3N4大。泊松比反映了材料在单轴形变情况下体积变化的大小,当泊松比等于0.25和0.5时分别代表中心力固体的上限和下限,当等于0.5时表示弹性形变中体积不发生变化。由表3所列的泊松比可以看出β-C3N4 (
比β-Si3N4小,这说明β-C3N4的不可压缩性比β-Si3N4大。泊松比反映了材料在单轴形变情况下体积变化的大小,当泊松比等于0.25和0.5时分别代表中心力固体的上限和下限,当等于0.5时表示弹性形变中体积不发生变化。由表3所列的泊松比可以看出β-C3N4 (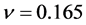 )和β-Si3N4 (
)和β-Si3N4 (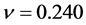 )原子间的结合力都不是中心力。β-C3N4和β-Si3N4的泊松比都小于0.25,说明形变时体积将发生较大的变化,且β-C3N4较β-Si3N4形变时体积的变化更大。同时,略小的泊松比表明材料抵抗剪切应变的过程中具有较好的稳定性。
)原子间的结合力都不是中心力。β-C3N4和β-Si3N4的泊松比都小于0.25,说明形变时体积将发生较大的变化,且β-C3N4较β-Si3N4形变时体积的变化更大。同时,略小的泊松比表明材料抵抗剪切应变的过程中具有较好的稳定性。
体弹模量代表材料化合键强度的平均值,而剪切模量体现材料受到外力作用时抵抗化合键改变的能力。因此,G/B的大小反映材料中键合定向性的程度。根据Pugh判据 [32] ,G/B > 0.57的材料整体显脆性;G/B < 0.57的材料整体显韧性。由表3可以看出,G/B的值:β-C3N4 (0.861) > β-Si3N4 (0.628),表明β-C3N4结构中化合键的定向性比β-Si3N4高,并且β-C3N4和β-Si3N4的G/B都大于0.57,因此它们都显脆性。
3.3. 硬度对比
硬度代表了固体材料抵抗弹性变形、塑性变形或者破坏的能力,它是表征材料力学性能的重要物理量。材料硬度的计算可根据晶体原子间不同的键合作用方式,采用不同的公式进行推算。本文采用以下三种计算方法分别对β-C3N4和β-Si3N4的硬度进行计算。
公式①
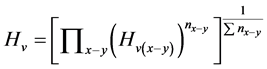 , (11)
, (11)
式中,
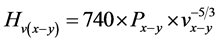 , (12)
, (12)
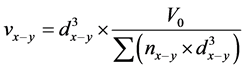 , (13)
, (13)
其中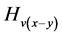 ,
,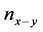 ,
,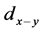 和
和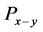 分别代表晶体由纯
分别代表晶体由纯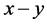 型键搭建时的硬度、原胞中
型键搭建时的硬度、原胞中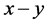 键的数目、
键的数目、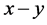 键的键长和键布居数,
键的键长和键布居数,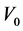 代表晶胞体积。
代表晶胞体积。
公式②
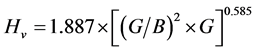 , (14)
, (14)
公式③
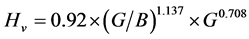 , (15)
, (15)
公式②和公式③中,G为剪切模量,B为体弹模量。
由公式① ② ③计算的β-C3N4,β-Si3N4和(C,Si)3N4的硬度如表4所示。
Table 4. The hardness (GPa) of β-C3N4 and β-Si3N4 calculated from the formula ① ② ③
表4. 由公式① ② ③计算的β-C3N4和β-Si3N4的硬度(GPa)
由表4可以看出,3种方法分别算出来的硬度相差无几,β-C3N4的硬度比β-Si3N4高1.5倍左右,这也是由于β-C3N4中的C-N键强度比β-Si3N4中Si-N键的强度更高所致。另外,表4也给出了β-C3N4和β-Si3N4的硬弹比(硬度/弹性模量),它的大小表征材料的耐磨性,材料的硬弹比越大,则耐磨性越好。数据表明,β-C3N4的耐磨性也比β-Si3N4更好。
4. 结论
我们采用基于密度泛函理论的第一性原理方法探索了C原子替换Si原子过程中体系能量最低的路径,即β-Si3N4转变为β-C3N4的最可能路径。在C原子替换Si原子过程中,晶胞体积逐渐减小,体系能量逐渐降低,体系在热力学上逐渐趋于稳定。另外,我们计算和对比了β-C3N4和β-Si3N4的弹性和硬度。根据六方结构晶体的弹性稳定性准则可以判定,β-C3N4和β-Si3N4的晶体结构都是稳定的。此外,β-C3N4的弹性模量E、体弹模量B和剪切模量G相比于β-Si3N4增大了1倍左右,硬度相比于β-Si3N4增大了1.5倍左右。而β-C3N4的泊松比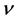 比β-Si3N4小,说明β-C3N4的不可压缩性比β-Si3N4大。G/B的值β-C3N4和β-Si3N4都大于0.57,且β-C3N4>β-Si3N4,表明β-C3N4和β-Si3N4都显脆性,但β-C3N4比β-Si3N4脆性更大。综上所述,β-C3N4具有稳定的晶体结构和较高的弹性和硬度,这有利于实际应用,但其脆性较大,因此还需要进一步研究和改进。
比β-Si3N4小,说明β-C3N4的不可压缩性比β-Si3N4大。G/B的值β-C3N4和β-Si3N4都大于0.57,且β-C3N4>β-Si3N4,表明β-C3N4和β-Si3N4都显脆性,但β-C3N4比β-Si3N4脆性更大。综上所述,β-C3N4具有稳定的晶体结构和较高的弹性和硬度,这有利于实际应用,但其脆性较大,因此还需要进一步研究和改进。
基金项目
国家自然科学基金资助(批准号:51601153)。