1. 引言
由于稀土金属–硅团簇作为一种新的功能材料的基元在新材料设计领域中有着极为重要的价值,所以稀土元素掺杂硅团簇受到人们的广泛关注[1] -[4] ,研究稀土金属原子掺杂硅团簇的结构和性质,不仅可以为稀土金属原子掺杂引起的硅团簇表面重构提供模型,而且可以为解释含有稀土金属原子硅团簇这类功能材料本身所具有的特殊性质如电磁性和光电性提供理论基础。
最近,Wang等人[5] 用密度泛函方法研究了CeSi6的结构和性质。虽然关于铈掺杂硅团簇在理论化学方面的研究目前还少见报道,但由于铈是最丰富的低毒的稀土元素,随着铈从早期的打火石、电弧碳棒、玻璃的脱色剂、澄清剂和研磨抛光剂应用到现代工业的光学玻璃、计算机芯片、合金材料、激光器、永磁材料、和汽车尾气净化催化剂等各个方面的广泛应用,铈掺杂团簇的研究必将受到人们的广泛关注,因为团簇作为物质体系从原子、分子向凝聚相的过渡状态本身就具有丰富、奇异的物理、化学性质,尤其在电、磁和光等方面的性质是其它类化合物难以比拟的,具有优越的应用前景[6] 。
2. 计算方法
采用B3LYP优化了CeSin (n = 1~8)的几何构型;分析了它们的频率,若出现虚频,进行Jahn-Teller修正,直到获得局域最小点;用NPA计算了基态结构的中原子的电荷和磁矩等性质。对硅原子采用cc-pVTZ基组,对铈原子采用Stuttgart RSC Segmented结合小核ECP28MWB赝势基组[7] [8] 。所有计算在Gaussian-09软件包[9] 中完成。优化的初始构型主要考虑三种构型,一种是“吸附结构”,即CeSin的构型可以看作是Ce原子吸附在Sin基态结构上而获得;一种是“取代结构”,即CeSin的构型可以看作是Ce原子取代Sin+1基态结构上的一个硅原子而获得;第三种是参照其它过渡金属或稀土金属掺杂硅团簇的基态结构自行设计的初始结构。铈的外层电子构型是4f15d16s2,Si和Si2的基态结构是3重态[10] ,所以CeSi和CeSi2在1或3或5重基态都是可能的,尽管如此,CeSin (n = 3~8)我们也考虑了单重态、3重态和5重态三种情况。
3. 结果与讨论
在图1至图8中给出了CeSin (n = 1~8)团簇的平衡构型和几何参数,其中:标数字的小球代表Si原子,未标数字的小球代表Ce原子;键长的单位为Å;ΔΕ为相对能差,单位为eV。
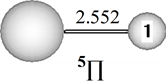
Figure 1. Ground state structure of CeSi
图1. CeSi的基态结构
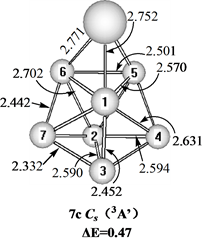
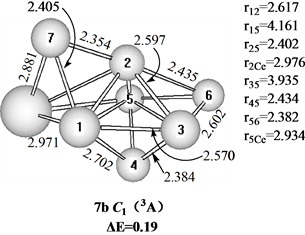
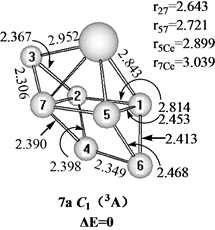
Figure 7. Three structures of CeSi7
图7. CeSi7的3种结构
3.1. CeSi
在B3LYP水平上,CeSi的基态结构是五重态(5Π),这一结果和Shaheen报导的CeSi基态结构的磁矩为4实验结果一致[11] 。当电子态为3Π、1Σ和7Σ时,它们的能量比基态分别高0.04、1.27和0.54 eV。
3.2. CeSi2
对CeSi2来说,可能的结构有直线形、弯曲形和三角形。但弯曲形的结构经优化最后成为三角形或直线形,所以在报导的CeSi2八种结构中没有弯曲结构。CeSi2的基态结构(2a)是具有C2v对称性的等腰三角形,电子态为3B1。异构体2b、2c和2d也是具有C2v对称性的等腰三角形,电子态分别为5B1、5A1和1A1,能量上它们分别比基态结构高0.90、1.02和1.12 eV。异构体2e和2f是具有C∞v对称性的线性结构,2g和2h是具有D∞h对称性的线性结构,它们的电子态分别为5Π、3Σ、5Σu和1Π,能量上它们分别比基态结构高1.92、2.76、2.85和3.44 eV。基态结构的Ce-Si键长为2.704 Å,比CeSi基态结构中的Ce-Si键长长了0.152 Å。
3.3 CeSi3
在图3中给出七个CeSi3的结构,其中四个(3a-3d)是三重态结构,三个(3e-3g)是单重态结构。具有C2v对称性的平面菱形(3a)被预测为基态结构,电子态为3B1,当电子态为3B2时(3b),它仅比基态的3B1在能量上高0.14 eV。异构体3e是具有C1对称性的四面体,能量上比基态的3a高0.67 eV。异构体3d也是具有C2v对称性的平面菱形结构,电子态为3A2,能量上它比基态的3a高0.78 eV。异构体3c可以看成是吸附结构,异构体3d可以看成是一个Ce原子取代Si4平面菱形基态结构[10] [12] 上的短对角线上的一个Si而获得,即取代结构,而异构体3a和3b即可以看成是吸附结构又可以看成是取代结构。对单重态的异构体3e-3g来说,能量上明显高于三重态的结构,它们分别比基态3a高1.27、1.28和1.66 eV。CeSi3基态结构中的Ce-Si键长比CeSi2基态结构中的键长约长0.062 Å。相对应的三个五重态结构没有画出,在5A1、5A和5B2电子态时其能量分别比基态大约高0.77、0.89和1.30 eV。
3.4. CeSi4
在图4中给出六个CeSi4的结构,三个(4a-4c)三重态的结构,三个(4d-4f)单重态的结构。具有Cs对称性和3A′电子态的4a结构被预测为是基态结构,它可以看成是一个Ce原子取代Si5三角双锥基态结构[10] [12] 上的一个Si原子而获得,即属于取代结构。异构体4b和4c属于吸附结构,即一个Ce原子分别吸附Si4平面菱形基态结构[10] [12] 的面上和棱上而获得。异构体4b和4c的对称性分别为C2v和Cs,电子态分别为3B1和3A″,能量上它们分别比基态4a结构高0.28和1.17 eV。相对应的三个单重态结构3d-3f在能量上分别比基态4a结构高1.06、1.94和2.23 eV。基态结构的三个Ce-Si键长中,两个相等的为2.799 Å,另一个为2.717 Å。相对应的两个五重态结构没有画出,在5A′、5B2和5A′电子态时其能量分别比基态大约高0.46、1.25和1.62 eV。
3.5. CeSi5
在图5中给出六个CeSi5的结构。如果用一个Ce原子取代Si6四角双锥基态结构[12] 上的一个Si原子,我们可以获得具有C2v对称性电子态分别为3B2和3A2的两个异构体,频率分析表明3A2电子态的异构体(5c)是一个局域最小点,3B2电子态的异构体是一个鞍点,因为它有一个b1模式的112i cm−1虚频,经Jahn-Teller修正,获得具有Cs对称性他3A″基态的5a结构,能量上它比3A2电子态的5b异构体约低0.55 eV。异构体5b可以看作是一个Ce原子吸附在Si5三角双锥基态结构[10] [12] 的面上而获得,具有Cs对称性和3Aʺ电子态,能量上它比基态的5a约高0.21 eV。异构体5d和5f是对应于5a和5c的单重态结构,单重态的5e异构体对应于三重态的5b结构,5b构型在单重态时有虚频,经Jahn-Teller修正,获得C1对称性的5e结构。能量上单重态的5d、5e和5f分别比基态的5a高1.08、1.31和1.69 eV,相对应的三个五重态结构没有给出,在5A″、5A1和5B2电子态时其能量分别比基态大约高0.82、0.84和1.08 eV。基态5a结构中的四个Ce-Si键长为2.790~2.899 Å。
3.6. CeSi6
在图6中给出五个三重态的CeSi6的结构。Wang等人[5] 用PBEPBE等密度泛函方法研究了CeSi6,报导了其基态结构为具有Cs对称性的扭曲的五角双锥。我们的结果和他们的结果是一致的(如图6a),具有3A基态,当在C2v对称性和3A2电子态的结构(图6b)时,其能量仅比3A′基态高0.03 eV。具有C1对称性的异构体6c属第三种类型的结构,能量上比基态的6a高0.33 eV。具有Cs对称性和3A′电子态的异构体6d,和6a一样是取代结构,即Ce原子取代Si7五角双锥基态结构[10] [12] 不同位置上的Si原子而获得,能量上6d结构6a基态结构高0.66 eV。具有Cs对称性3Aʺ电子态的6e异构体可以看作是一个Ce原子吸附在Si6四角双锥基态结构[12] 的面上而获得,能量上它比6a基态结构高0.70 eV。我们也计算了单重态和五重态的异构体,只是能量上比基态结构高很多,所以在图中未给出。
3.7. CeSi7
在图7中给出三个三重态的CeSi7的结构。具有C1对称性的7a结构可以看作是一个Ce原子取代Si8双顶四角双锥基态结构[10] 的一个Si原子而获得,具有C1对称性的7b异构体可以看作是一个Si原子吸附在CeSi6基态结构的一个面上而获得,具有Cs对称性和3A′电子态的7c异构体可以看作是一个Ce原子吸附在Si7五角双锥基态结构[10] [12] 的一个面上而获得,能量上,基态的7a结构比7b和7c异构体分别稳定0.19和0.47 eV。我们也计算了单重态与五重态的异构体,只是能量上比基态结构高很多,所以在此没有给出。
3.8. CeSi8
在图8中给出五个CeSi8的结构。具有C2v对称性和3B1基态的8a结构即可以看作是一个Ce原子取代Si9扭曲的双顶五角双锥基态结构[10] 的一个Si原子而获得,也可以看作是一个Ce原子吸附在Si8双顶四角双锥基态结构[10] 的一个面上而获得。异构体8c和8d属于取代结构,它们具有Cs对称性,电子态分别为3A′和3A″,能量上它们比8a分别高0.58和1.07 eV。具有C1对称性和三重态的8b异构体属于第三种类型,能量上它比基态的8a结构稍稍高0.11 eV。我们只给出了一个单重态的异构体8e,它具有C2v对称性,电子态为1A1,能量上比基态8a约高1.00 eV。另外,单重态和五重态的异构体能量比较高,稳定性较差。由于Si原子较少所以不能形成囊形结构,当用“囊形结构”作为初始结构优化时,Ce原子最后析出到团簇表面。
3.9. 断裂能
从CeSin (n = 1~8)团簇上断裂一个Ce原子(DE1 = E(Sin) + E(Ce) − E(CeSin))和一个Si原子(DE2 = E(Sin-1) + E(Si) − E(CeSin))的断裂能分别在图9中给出。从断裂能中可知它们的相对稳定性。从图9中可知DE1和DE2的变化趋势一致;CeSi2、CeSi5和CeSi8的断裂能是局域最大,说明它们比较稳定,而CeSi4和CeSi7断裂能是局域最小,所以稳定性差,这也说明在裸硅团簇中Si4和Si7是比较稳定的,该结果和以前的研究结果一致[10] [12] 。
3.10. 电子性质和磁矩
为了进一步的了解CeSin (n = 1~8)基态团簇中Si原子与Ce原子之间的相互作用,我们用NPA计算了基态结构中原子的电荷、磁矩和偶极矩等性质,分别在表1和表2中给出。从表1中可知Ce原子在CeSin (n = 1~8)基态团簇中的电子排布是6S0.16~0.894f1.14~1.245d1.36~2.156p0.05~0.18,和Ce原子6S24f15d16p0比较可知,当Ce原子掺杂硅团簇时,6s轨道上的电子数在减小,4f和5d轨道上的电子数在增加,随着Si原子数目的增多,6s轨道上的电子的数目单调减小,4f轨道变化不明显,5d轨道显著增加,即6s轨道上的电子一部分主要转移到了5d轨道上。Ce原子在CeSin (n = 1~8)基态团簇中的电荷始终为正值(从0.37 a.u.到0.68 a.u.),说明Ce原子在CeSin (n = 1~8)结构中是电子供体,电荷从Ce迁移到Sin团簇。偶极矩可以探测电荷分布,对于CeSin (n = 1~8)基态团簇来说,由于电子从Ce原子迁移到Sin团簇,产生了偶极矩。其中,CeSi8的偶极矩最大,为9.86D,CeSi的偶极矩最小,为4.67D。CeSi偶极矩最小的原因是Ce与Si之间电荷迁移的少以及正负电荷中心间的距离也短,虽然在CeSi8中Ce与Si8团簇之间电荷迁移的也不是很多,但在CeSi8中正负电荷中心间的距离很大,从而产生了最大的偶极矩。从表2中可知,除了CeSi的总磁矩为4之外,其它团簇的总磁矩为2。从Ce原子的局域磁矩可知,CeSin团簇的总磁矩主要是由掺杂的Ce原子所提供,尤其是CeSi3,总磁矩全部来源于Ce原子。
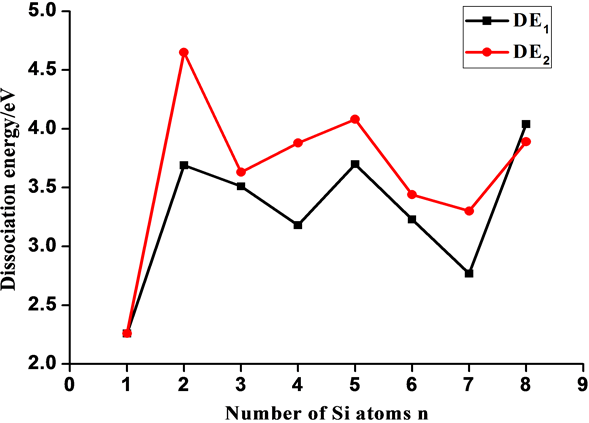
Figure 9. Dissociation energy versus the number of atoms n for the ground state structure CeSin
图9. CeSin基态的断裂能随Si原子数目变化

Table 1. The electron configuration and charge (in a.u.) of Ce atom and dipole moment (in Debye) for CeSin (n = 1 - 8) clusters
表1. CeSin (n = 1~8)团簇中的Ce原子的电子排布、电荷和偶极矩
Table 2. Magnetic moment (μB) of 6s,4f, 5d, and 6p orbitals for Ce atom, total magnetic moment (μB) of the Ce atom, and total magnetic moment of the lowest-energy of CeSin clusters
表2. 基态CeSin团簇中Ce原子的6s,4f, 5d, 6p轨道磁矩,Ce的总磁矩以及CeSin团簇的总磁矩
4. 结论
采用B3LYP结合稀土元素小核ECP28MWB基组,系统地研究了CeSin (n = 1~8)体系的几何构型、电子结构、断裂能、磁性和偶极矩等性质。CeSin基态可以看作是Ce取代Sin+1上的一个Si原子所获得,除了CeSi基态是5重态之外,其它物质是3重态。预测的从CeSin(n = 1~8)团簇上断裂一个Ce原子的断裂能分别为2.26eV,3.69 eV,3.51 eV,3.18 eV,3.70 eV,3.23 eV,2.77 eV,4.04 eV;断裂能表明CeSi2、CeSi5和CeSi8相对比较稳定,而CeSi4和CeSi7相对稳定性较差。电荷分布表明,在CeSin中,Ce原子是电子供体,Sin团簇是电子受体,Ce原子的4f电子变化不大,主要失去6s电子,部分进入5d轨道,部分进入Sin团簇。预测的偶极矩分别为4.68D,7.76D,7.60D,9.25D,9.06D,7.08D,7.56D,9.86D; CeSi的偶极矩最小,CeSi8的偶极矩最大。CeSin团簇的磁矩主要由掺杂的Ce原子所提供,Ce原子在CeSin团簇中的局域磁矩分别为2.74μB,1.59μB,2.01μB,1.41μB,1.42μB,1.86μB,1.35μB,1.52μB。
基金项目
该课题得到国家自然科学基金资助(No.21263010)。