摘要:
通过文献研究发现鹅不食草具有明显的抗炎活性,但其药效物质基础和作用机制尚不清楚。因此利用分子对接技术对鹅不食草中具有抗炎作用的化学成分进行虚拟筛选。在TCMSP数据库中筛选出鹅不食草的化学成分组成配体数据库,选择4个与抗炎活性密切相关的蛋白靶点组成受体数据库,用SYBYL软件进行分子对接。通过对接打分函数(Total Score ≥ 5)筛选出6个活性成分,并分析了蛋白复合体的相互作用。本研究初步筛选了鹅不食草抗炎的活性成分和关键靶点,为鹅不食草的进一步研究提供了参考。
Abstract:
Through literature research, it was found that Herba centipedae has obvious anti-inflammatory activity, but its pharmacodynamic substance basis and mechanism of action are unclear. There-fore, the molecular docking technology was used to screen the chemical components in Herba centipedae with anti-inflammatory effects. In the TCMSP database, the chemical composition of Herba centipedae was selected to form a ligand database, 4 protein targets closely related to an-ti-inflammatory activity were selected to form a receptor database, and molecular docking was performed with SYBYL software. Six active ingredients were screened by the docking scoring function (Total Score ≥ 5), and the interaction of protein complexes was analyzed. In this study, the anti-inflammatory active ingredients and key targets of Herba centipedae were preliminarily screened, which provided a reference for its further research.
1. 引言
炎症是一种基本的病理生理过程,是机体在受到致炎因子刺激后产生的一种防御反应,其特点是会产生大量炎性介质和促炎因子,比如组胺、前列腺素E2 (PGE2)、一氧化氮(NO)、白介素1 (IL-1)和肿瘤坏死因子-α (TNF-α)等 [1]。炎症反应是一些疾病的发病基础,如不及时干预治疗可能会造成严重后果 [2]。鹅不食草为菊科植物石胡荽(Centipeda minima (L.) A. Br. et Aschers.)的全草,被《中国药典》收载,其味辛、性温,具有发散风寒、通鼻窍、止咳、解毒、止痒等功效,传统上用于治疗鼻炎、疟疾、哮喘等,广泛分布于东亚和东南亚地区 [3]。鹅不食草中含有多种化学成分,如挥发油类、甾体类、黄酮类、萜类、有机酸等 [4]。近年来研究表明其药理活性主要包括抗炎、抗肿瘤等 [5],但在鹅不食草中,哪些成分具有抗炎活性,以及其具体抗炎机制仍不清楚。计算机辅助药物设计(Computer-Aided Drug Design, CADD)是借助计算机原理,通过模拟药物与受体生物大分子的相互作用,预测其亲和力,实现基于结构的药物设计的一种重要方法 [6],其应用极大地加快了新药研发的速度与效率,已成为现代药物研发的常规方法之一。而分子对接技术则是计算机辅助药物分子设计的重要手段之一 [7]。本研究即以分子对接技术为研究方法,对鹅不食草中所含成分进行研究,进一步明确鹅不食草中主要抗炎药效物质,并阐明其药理作用机制,也为中药多靶点研究提供新的参考。
2. 材料与方法
2.1. 程序系统
所有研究及计算工作均在SYBYL-X2.0 Windows版本中的Surflex-dock模块中完成。
2.2. 靶点的准备
本研究的靶点均从Protein Data Bank (PDB)数据库(http://www.rcsb.org/)中获取,根据文献 [6] [8] [9] [10],选取了4个蛋白进行研究,是目前抗炎药物设计的重点靶标,因此在本研究中选为代表靶点。在PDB数据库中下载这4个蛋白的晶体结构,具体蛋白靶点见表1。将下载的靶蛋白导入SYBYL软件中,进行蛋白晶体前处理,提取原配体,删除水分子以及非相关的蛋白质构象,对非完整的氨基酸残基进行补充,加极性氢原子,以原配体分子位置为中心,设置半径为5埃,将此范围作为活性口袋,其他参数为默认值。
2.3. 配体的准备和处理
以“鹅不食草”为关键词,在TCMSP (http://lsp.nwu.edu.cn/index.ph)数据库检索出鹅不食草所含的化学成分及其药动学参数。挑选出口服生物利用度(oral bioavailability, OB) ≥ 30%,类药性(drug-likenesses, DL) ≥ 0.18的化合物共21个,组成化合物数据库。从PubMed (http://pubchem.ncbi.nlm.nih.gov/)中下载相应化合物的平面结构,未能在PubMed下载的化合物用ChemDraw绘制结构式,保存为mol2格式,化合物具体名称见表2。在sybyl-X2.0中创建配体小分子数据库,将这些化合物导入后进行能量最小化处理,使每个化合物有最稳定的构象,最后导入数据库。

Table 1. Target protein information
表1. 靶点蛋白信息
Table 2. Information about compounds contained in Herba centipedae
表2. 鹅不食草中所含化合物的相关信息
3. 结果
按默认参数进行分子对接后,分别得到每个化合物与4个靶点蛋白的对接得分Score值,Score值是用−log(KD)表示的,由于RT ln(KD) = G (配体与受体的结合自由能),因此G值与Score值成反比。结合自由能越小,配体与受体结合的稳定性就越高,Score值越大,G值越小,配体与受体的结合越稳定。本研究将每个化合物与4个蛋白靶标对接后的Score加和,并根据总分值从高到低排列,具体结果见表3。由打分结果可知,总分高于阈值的化合物共6个,具体结构见图1。内在配体与靶点分子对接结果见图2。
从表3可知,鹅不食草的21种化合物与3nt1、1m9t、2zoq、4l8m进行分子对接后,得分最高的化合物是化合物17金色酰胺醇酯;单个靶点得分最高的化合物除化合物17外还有化合物12谷甾醇、化合物10蒲公英甾醇棕榈酸酯。进一步分析化合物17金色酰胺醇酯与3nt1、1m9t、2zoq、4l8m蛋白相互作用模式。由图3~6可以看出化合物17与3nt1的Arg216、Tyr134和Tyr147氨基酸残基形成氢键,与Leu238、Asp239、His133等5个氨基酸残基有疏水相互作用;与1m9t的Trp366和Met368氨基酸残基形成氢键,与Gly196、Val346、Cys194等9个氨基酸有疏水相互作用;与2zoq的Arg87氨基酸残基形成氢键,与Lys220、Val205、Arg189等10个氨基酸残基有疏水相互作用;与4l8m的Gly110氨基酸残基形成氢键,与Asp112、Leu171、Leu167等10个氨基酸有疏水相互作用。如图7所示,3nt1靶点得分最高的化合物是化合物12谷甾醇,其与蛋白靶点的Leu224氨基酸残基形成氢键,与Leu145、Arg216、Ser143等7个氨基酸残基有疏水相互作用;2zoq靶点得分最高的化合物是化合物10蒲公英甾醇棕榈酸酯,其与蛋白靶点的Val205氨基酸残基形成氢键,与Glu203、Arg87、Thr202等7个氨基酸残基有疏水相互作用,见图8。正是因为这些残基与配体的相互作用,增强了配体与受体的结合能力,从而使该配体化合物的活性提高。
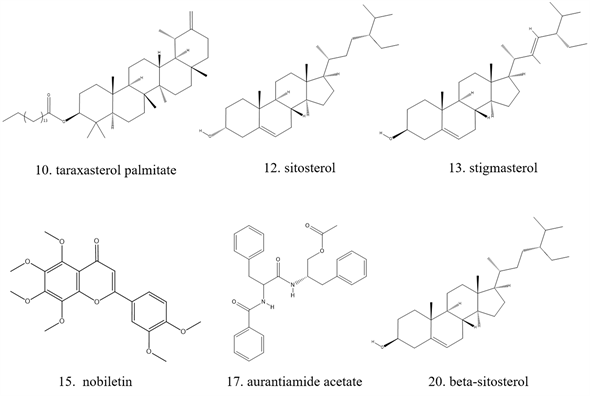
Figure 1. Active ingredients in Herba centipedae
图1. 鹅不食草所含活性成分

Figure 2. The molecular docking of the internal ligand and each target molecule
图2. 内在配体与各靶点分子对接图
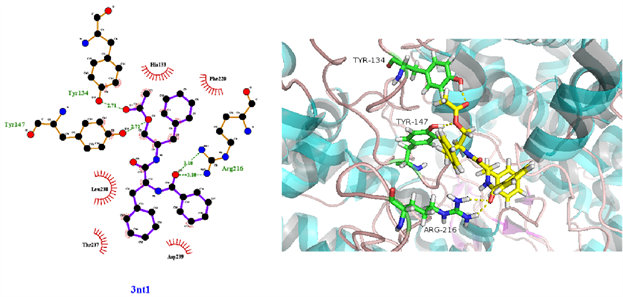
Figure 3. The molecular docking of 3nt1 and Compound 17
图3. 3nt1与化合物17对接

Figure 4. The molecular docking of 1m9t and Compound 17
图4. 1m9t与化合物17对接
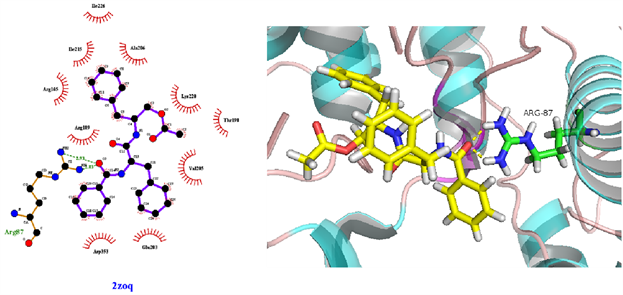
Figure 5. The molecular docking of 2zoq and Compound 17
图5. 2zoq与化合物17对接
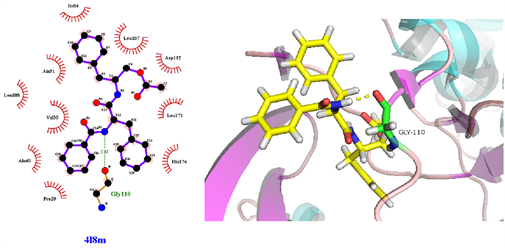
Figure 6. The molecular docking of 4l8m and Compound 17
图6. 4l8m与化合物17对接

Figure 7. The molecular docking of 3nt1 and Compound 12
图7. 3nt1与化合物12对接
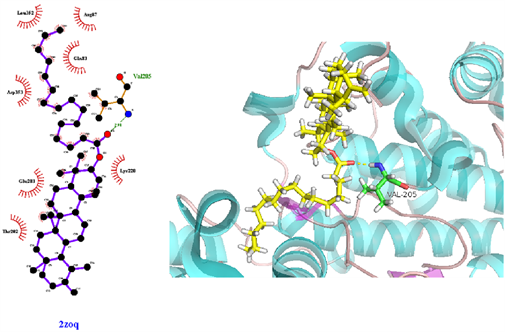
Figure 8. The molecular docking of 2zoq and Compound 10
图8. 2zoq与化合物10对接
4. 讨论
由于中药成分复杂,其药理作用具有多靶点、多作用的特点,很难用传统方法详细阐述其药效学基础和药物作用机理 [11]。计算机虚拟筛选技术在分析有效成分和药物开发方面显示出其独特的优势。利用计算机辅助药物设计来解释特定小分子的反向药理机制已引起越来越多的关注,这为阐明天然产物的作用靶点和机制提供了一种新的方法,也为药物研究提供了新思路。本研究应用计算机虚拟筛选技术,以炎症信号通路的关键靶点COX-2、iNOS、ERK1、p38为靶蛋白,对鹅不食草中21个化学成分进行分子对接筛选,综合评价对接结果,筛选出6个活性较好的化合物。在筛选出的候选化合物里,其中一些化合物的生物活性已被报道。如李等 [12] 研究证明金色酰胺醇酯在脂多糖诱导的RAW264.7细胞炎症模型中显示出一定的抗炎活性。张等 [13] 证明化合物15川陈皮素对二甲苯所致小鼠耳肿胀具有明显的抑制作用。Gabay等 [14] 研究表明化合物13豆甾醇抑制多种炎症介质和基质降解。Tan等 [15] 报道β-谷甾醇能明显降低组胺诱发的小鼠毛细血管通透性增加,具有一定的抗炎的活性,这些报道为本研究结果提供了有力支撑。其中化合物10蒲公英甾醇棕榈酸酯在结果中也表现出较好活性,但其作用靶点及机制未见报道,程等 [16] 证明蒲公英甾醇可以通过调节p38和ERK 1/2 MAPKs的磷酸化水平抑制iNOS和COX-2的表达来发挥体外抗炎作用,而分子对接结果显示其侧链连接了棕榈酸之后仍有较强活性。从药物设计的角度讲,很多药物成酯后可以消除一些感官上的不良反应,提高药物安全性,这给下一步工作提供了新的研究思路,同时也说明分子对接技术在虚拟筛选中药活性方面具有一定的可行性并有助于找到所需要的先导化合物,为鹅不食草的开发利用提供了指导和借鉴。
本研究首次将分子对接应用到鹅不食草的抗炎化合物的筛选,这相对于传统筛选,节约了大量时间和精力。当然,此次研究只采用分子对接技术进行初步筛选是不够的,下一步还将结合药效团模型和分子动力学等其他技术进行综合研究和评价。利用这些计算机模拟方法探讨中药的作用机制,不仅可以弥补药理学实验方法的缺陷,而且能在中药新药研究领域引进新的理念和先进的研究方法,为现代中药研究提供新思路和新方法。
NOTES
*通讯作者。