1. 引言
百草枯是目前世界范围内使用最广泛的除草剂之一,在我国的使用也非常普遍。近年来,国内关于百草枯中毒的报导屡见不鲜,引发医疗、生产、使用、管理部门和公众对于百草枯安全问题的一致关注。目前世界上尚没有能解百草枯的特效药,资料显示,全国百草枯中毒的救治率低于20%,口服20%水剂后死亡率高达95%,甚至有口服1毫升即致死的先例。在临床上误服百草枯后会出现各脏器衰竭的症状,特别是肺部出现呼吸交迫综合症后很难挽救患者生命。百草枯在农村是可以在农药店都能买到的常见农药,对于情绪不稳定的人和好奇心较强的儿童来说,百草枯成为了对人威胁很大的毒药 [1]。催吐剂:必需的“后悔药”为了防止误服百草枯而造成悲剧,2004年起颁布的百草枯的母液和制剂的国家标准,强制要求企业必须在百草枯中加入一定比重的催吐剂。在这两份国家标准中都规定了:百草枯阳离子与2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮(又名PP796,三氮唑嘧啶酮)。2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮较百草枯吸收快,持续期长,能够作用于人的脑呕吐中心,且对肠胃无刺激。因而,2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮被公认为理想的催吐剂 [2]。
三氮唑嘧啶酮合成方法主要有三种:1) 以N-正丙基-1H-1,2,4-三唑-3,5-二胺为起始原料,经过缩醛保护,与3-甲氧基-2-甲基丙烯酸甲酯反应,经脱保护得到目标产物 [3] [4] [5];2) 以1,2,4-三唑-3,5-二胺为起始原料,先与2-甲基丙烯酸甲酯反应,所得产物再与溴丙烷反应得到目标产物 [6] [7] [8];3) 嘧啶直接催化闭环法等 [9] [10]。综合三种路线的优缺点,本文选择了下列合成路线:N-正丙基-1H-1,2,4-三唑-3,5-二胺为起始原料,经过苯甲醛保缩醛护,在碱催化下与3-甲氧基-2-甲基丙烯酸甲酯反应,经脱保护得到目标产物并回收苯甲醛等3步的合成路线,各步收率均较理想,分别达96%以上,90%以上,95%以上,该路线原料易得,合成总收率经优化提高到82%以上。经优化后的工艺,收率高,“三废”少,所使用的溶剂均为单一的甲苯溶剂,苯甲醛可以回收套用至第一步化合物I的合成,酸解后直接结晶无需调pH值,所得母液可以直接用于下一步的酸解脱保护,经实验母液可以直接套用6次,套用6次后产品质量出现下降,将次母液蒸馏后补加盐酸后再继续使用。大幅降低“三废”的排放,产品质量纯度大于99.5%,高于市场平均水平。
2. 实验
2.1. 原料和仪器
N-正丙基-1H-1,2,4-三唑-3,5-二胺(工业,南京恰风和医药科技有限公司),3-甲氧基-2-甲基-丙烯酸甲酯(工业,上海复和化学科技有限公司),甲醇钠(工业,浙江先锋科技股份有限公司),冰醋酸(AR,麦克林试剂有限公司),甲苯(工业,浙江瓯华化工进出口有限公司),精制盐酸(36%,台州市液体化工有限公司),苯甲醛(工业,江苏顺丰化工有限公司),焦亚硫酸钠(AR,麦克林试剂有限公司),对甲氧基苯酚(AR,麦克林试剂有限公司)。
高效液相色谱仪(安捷伦1260),核磁共振色谱(BrukerAvance III 400MH)。
2.2. 反应过程
2.2.1. 2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮合成路线
2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮的合成路线详见图1。
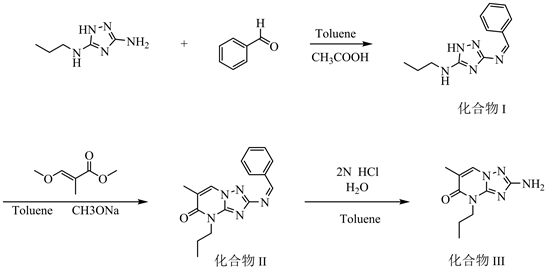
Figure 1. The synthetic route of 2-Amino-6-methyl-4-propyl-4,5-dihydro[1,2,4]triazolo[1,5-a]pyrimidin-5-one
图1. 2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮的合成路线
2.2.2. N-苄基亚胺-N-丙基-1H-1,2,4-三唑-3,5-二胺(化合物I)的制备方法
室温下,向具有回流分水装置的5000 ml四口烧瓶中投入甲苯2500 g,搅拌下投入固体N-正丙基-1H-1,2,4-三唑-3,5-二胺(二胺) 500.0 g,白色固体不溶,100 g甲苯洗涤投料口;继续投入401 g苯甲醛(新的苯甲醛或回收的苯甲醛),四口烧瓶内反应体系变为黄色,投料过程中不放热。升温50~60℃ (部分固体溶解),加入乙酸3.0 g,50 g甲苯洗涤器具后投入烧瓶。升温回流分水,搅拌转速不低于300 rpm,回流分水6 h,无水分出后取样检测,要求N-正丙基-1H-1,2,4-三唑-3,5-二胺 < 0.5%,化合物I的选择性 > 98.5%检测反应结束,降温至T < 50℃,进行下一工序的操作。1H NMR (CDCl3, 400 MHz) δ:9.11 (s, 1H),7.95~7.98 (m, 2H),7.50~7.55 (m, 3H),6.87 (s, 1H),3.122 (t, 2H),1.540 (m, 2H),0.885 (t, 3H)。HRMS:229.291。
2.2.3. 2-苄基亚胺-6-甲基-4-丙基-4H-[1,2,4]-三唑-[1,5-α]嘧啶-5-酮(化合物II)的制备方法
维持50℃以下,搅拌下投入对甲氧基苯酚1.2 g和固体甲醇钠60.5 g,投入经过滤的3-甲氧基-2-甲基丙烯酸甲酯512.5 g。升温至107~115℃,以约80~100 g/h的速度蒸馏含甲醇甲苯,同时补加等量的甲苯。反应15 h后,除甲醇钠外固体基本溶清,溶液呈暗红色。HPLC检测反应,要求化合物I < 1%。检测反应结束,降温至80℃。加入自来水800 g,搅拌30 min,静置分层。有机层继续降温,70℃有大量固体析出,在此温度保温结晶3 h。保温结束,继续降温至10~15℃,保温结晶3 h。过滤,滤饼用冰甲苯100 ml淋洗,抽干,真空干燥得到化合物II 925.3 g,两步总收率88.43%。1H NMR (CD3Cl, 400 MHz) δ:9.27 (s, 1H),7.89 (m, 2H),7.39 (m, 3H) 6.14 (s, 1H),4.15 (m, 2H),1.71 (m, 2H),1.66 (s, 3H),0.819 (t,3H)。HRMS:295.352。
2.2.4. 2-氨基-6-甲基-4-正丙基-1,2,4-三唑[1,5-a]并嘧啶-5-酮(化合物III)的制备方法
在具有减压蒸馏和分水设备的5000 ml四口烧瓶中投入化合物II 900.0 g,工艺水2625.0 g搅拌,下滴加入盐酸776.0 g,微负压下加热(−0.01~−0.02 MPa),温度70~75℃蒸出含苯甲醛的水溶液,水层流回烧瓶内,苯甲醛的甲苯溶液采用液相外标法检测含量后直接用于化合物I的制备。水解16 h,溶液澄清,取样检测HPLC,要求化合物II < 0.3%。反应结束后,降温结晶,约60~65℃开始有固体析出,继续降温至10~15℃,加入3 g焦亚硫酸钠抗氧剂,保温结晶3 h。过滤,所得母液直接用于下一批水解反应,循环套用6次后作废液处理;所得滤饼用纯化水淋洗两次,每次200 g,洗涤流出水的pH值6~7。真空干燥16 h,真空度不低于−0.085 MPa,温度55 ± 2℃。得到化合物III 601.2 g (母液套用6次的平均得量,平均收率为95.2%),HPLC纯度不低于99.7%。1H NMR (CD3Cl, 400 MHz) δ:7.7 (s, 1H),5.91 (s, 2H),4.21 (m, 2H),2.13 (s, 3H),1.79 (m, 2H),0.91(t, 3H)。HRMS:207.240。
3. 结果与讨论
3.1. 苯甲醛用量对化合物I反应的影响
根据HPLC分析结果(详见表1 HPLC分析结果部分)可知,苯甲醛的用量对反应二胺的转化影响较为明显,当苯甲醛低于1.07倍时反应转化不符合转化要求,高于1.07倍反应转化变化不够明显。经过实验发现苯甲醛超过1.07倍时对后面产品的质量影响较大,分析原因为大过量的苯甲醛与3-甲氧基-2-甲基丙烯酸甲酯和化合物I在碱性条件下发生较为复杂的副产物,虽然HPLC检测纯度时无影响,但会对化合物III产生明显的质量和收率影响。故本步骤反应需要严格控制苯甲醛的用量,特别是回收苯甲醛中含有甲苯,故需要对回收苯甲醛的用量需要准确的计算。建议大生产时集中处理回收苯甲醛,小试验证其中的含量再用于车间套用。

Table 1. Effects of nitrogen flow rate on Intermediate II reaction
表1. 苯甲醛用量对化合物I反应的影响
3.2. 催化剂和反应温度对化合物I制备反应的影响
实验过程中实验了对甲苯磺酸、甲磺酸、盐酸。冰醋酸,冰醋酸的催化效果最好,其他酸酸性较强催化产生的杂质较多,对反应都不利。反应溶剂考察了甲苯、二甲苯、正庚烷、四氢呋喃的回流条件下,正庚烷和四氢呋喃转化率达不到工艺要求,残留的二胺在10~15%之间,甲苯和二甲苯回流分水下都能达到工艺要求,仅回流不分水二胺的残留在3%~5%之间,无法达到反应终点,分析原因为水分高了反应达到平衡无法继续转化,必须将反应生成的水移除才能将反应进行的较为彻底。
3.3. 化合物II制备过程的影响因素
催化剂方面本文研究了颗粒状无水碳酸钾、粉末状无水碳酸钾、三乙胺、甲醇钠、醋酸钠。颗粒状碳酸钾转化较慢,反应30 h转化率仅达到60%左右,继续保温反应杂质增加,产物反而降低,分析原因为3-甲氧基-2-甲基丙烯酸甲酯时间久分解导致反应无法继续进行,并生成其他杂质。粉末状无水碳酸钾的反应速度比颗粒状碳酸钾要快,反应30 h时,化合物I < 3.0%。三乙胺催化反应选择性较差,产物的含量低于40%。醋酸钠的催化效果也较差,转化率和选择性都不理想。固体甲醇钠催化的选择性和转化率都比较理想,转化率在98%以上,选择性在95%左右,反应16 h即可完成。
实验过程发现边蒸甲苯边反应的转化率要比回流条件下的转化率高20%以上,蒸出反应生成的甲醇有利于反应的进行,要及时补加与蒸出等量的甲苯。
3.4. 母液套用次数对化合物III的收率和质量的影响
由表2可以看出,未套用母液的批次,收率仅达到86.4%,套用的收率可以达到96%以上,根据实验结果母液套用六次得到的产品的质量和收率均符合工艺要求,套用第七次化合物II不能完全转化质量呈明显下降的趋势,第八次套用就更加明显。所以母液套用六次必须要停止套用,我们对套用六次后的母液进行浓缩,调碱发现母液中并无物料析出,证明化合物III在盐酸溶液中的溶解度并不大,且不会生成盐酸盐。
Table 2. Effect of the times of motherliquor applications on the yield and quality of compound III
表2. 母液套用次数对化合物III的收率和质量的影响
3.5. 水解温度对化合物III的影响
本文对化合物II的水解温度进行了实验研究,60~70℃时随着反应的进行化合物II还未溶清,化合物III就已经析出来,导致后期反应进行的非常慢,且最终化合物剩余20%左右,反应就无法进行下去。高于80℃时,反应产生的杂质会变大,不适合做反应条件。
4. 结论
本文以N-正丙基-1H-1,2,4-三唑-3,5-二胺和苯甲醛为原料,经缩醛保护得到化合物I,再经闭环反应得到化合物II,化合物II脱保护得到目标产物。对各步骤的反应条件进行不断的优化摸索,反应的收率比较高,产品质量好,为以后工业化做好了技术储备。综合各工艺条件,化合物I的反应条件最终选择在甲苯为溶剂、冰醋酸催化、苯甲醛过量1.07倍,回流分水;化合物II的最优条件是甲苯做溶剂、甲醇钠催化常压下及时将生成的甲醇带出;化合物III的最优条件是70~75℃反应、微负压下蒸出脱下来的苯甲醛,反应完全后直接降温结晶,母液再套用六次。三步的收率分别达到96%,90%,95%;总收率达到82%以上。