摘要: 目的:探讨儿童不同遗传代谢性肝病的临床特点及病理诊断价值。方法:回顾性分析2018年1月至2022年6月重庆医科大学附属儿童医院行超声引导下经皮肝穿刺活检、临床诊断考虑遗传代谢性肝病且具备组织学资料的215例患儿的病因,总结最终诊断为遗传代谢性肝病患儿的临床特点及病理诊断价值。结果:215例中诊断不明67例,非遗传代谢性肝病67例,遗传代谢性肝病81例。64/81例病理学诊断:33例糖原累积症(GSD)、24例肝豆状核变性(WD)、2例脂质代谢障碍性疾病、1例尼曼–匹克病(NPD)、1例Alagille综合征(ALGS)、1例进行性家族性肝内胆汁淤积症(PFIC)、2例病因未明遗传代谢性肝病(IMLD)。不同遗传代谢性肝病的转氨酶有不同程度升高,36例GSD临床表现为肝脏肿大、低血糖、生长发育迟缓、乳酸升高等;34例WD多有血清铜蓝蛋白降低和24小时尿铜升高;2例脂质代谢障碍和1例NPD均表现为肝脏肿大、低血糖和高血脂;3例ALGS表现为不同程度的胆汁淤积;2例PFIC表现为黄染、肝脏肿大、总胆红素和胆汁酸升高;2例病因未明IMLD临床表现与各型IMLD有重叠;1例Citrin蛋白缺乏症临床表现为黄染、胆汁淤积及肝脾大。部分肝病有特征性组织病理改变,如GSD的植物细胞样肝细胞和细胞器边聚现象;WD的肝细胞偶见糖原化核和线粒体改变;脂质代谢障碍的肝细胞肿胀和Kupffer细胞增多;NPD的大量空泡结构,空泡内有髓样小体;ALGS的小胆管缺乏。结论:儿童遗传代谢性肝病可在任意年龄起病,转氨酶升高、生长发育迟缓、肝脾肿大、反复发作的胆汁淤积等是常见的临床表现,光镜结合电镜的组织学检查可以提高儿童遗传代谢性肝病的检出率,但最终的病理诊断或解释需结合患儿的症状和体征、实验室检查、家族史、影像学以及遗传学分析。
Abstract:
Objective: To explore the clinical characteristics and pathological diagnostic value of different inherited and metabolic liver diseases in children. Methods: The etiology of 215 children with inherited metabolic liver disease and histopathological data who underwent ultrasound-guided percutaneous liver biopsy in the affiliated Children’s Hospital of Chongqing Medical University from January 2018 to June 2022 were reviewed, to summarize the clinical characteristics and pathological diagnostic value of children with inherited metabolic liver disease. Results: Among the 215 cases, there were 67 cases of unknown diagnosis, 67 cases of non-inherited metabolic liver disease and 81 cases of inherited metabolic liver disease. 64/81 cases of pathological diagnosis: There were 33 cases of glycogen storage diseases (GSD), 24 cases of Wilson’s disease (WD), 2 cases of lipid metabolic disorders, 1 case of Niemann-Pick disease (NPD), 1 case of Alagille syndrome (ALGS), 1 case of progressive familial intrahepatic cholestasis (PFIC) and 2 cases of unknown inherited metabolic liver disease (IMLD). Transaminase in different genetic and metabolic liver diseases increased in varying degrees. 36 cases of GSD showed hepatomegaly, hypoglycemia, growth retardation and increased lactic acid, 34 cases of WD showed decreased serum ceruloplasmin and elevated 24-hour urinary copper, 2 cases of lipid metabolism disorder and 1 case of NPD showed hepatomegaly, hypoglycemia and hyperlipidemia, and 3 cases of ALGS showed different degrees of cholestasis. PFIC showed yellow staining, hepatomegaly, elevated total bilirubin and bile acid in 2 cases. The clinical manifestations of 2 cases with unknown etiology of IMLD overlap with various types of IMLD, and clinical manifestations of yellow staining, cholestasis and hepatosplenomegaly in 1 case of Citrin protein deficiency. Some liver diseases have characteristic histopathological changes, such as edge aggregation of plant cell-like hepatocytes and organelles in GSD, occasional changes of glycogen nuclei and mitochondria in hepatocytes of WD, swelling of hepatocytes with disturbance of lipid metabolism and increase of Kupffer cells, a large number of vacuolar structures of NPD with myeloid bodies in vacuoles, and lack of small bile ducts in ALGS. Conclusion: Children with hereditary and metabolic liver disease can begin at any age. Elevated transaminase, growth retardation, hepatosplenomegaly and recurrent cholestasis are common clinical manifestations. Histological examination of light microscope combined with electron microscope can improve the detection rate of inherited metabolic liver disease in children, but the final pathological diagnosis or explanation should be combined with symptoms and signs, laboratory examination, family history, imaging and genetic analysis.
1. 引言
儿童遗传代谢性肝病(inherited metabolic liver disease, IMLD)是一种广泛而多样的遗传疾病,病因复杂且种类繁多,既往因诊断困难而报道较少,但随着临床诊疗技术的提升和认知水平的提高,IMLD已成为一类常见的儿童肝病,国内有研究报道代谢相关性疾病(89/194, 45.88%)在儿童非病毒性肝病中最多 [1] 。1883年Paul Ehrlich在德国首次进行经皮肝活检,此后随着技术的进步,肝穿刺活检在临床中得到广泛应用 [2] ,遗传代谢性肝病的组织病理学显示不同程度的肝细胞损害及相应的病理改变,有助于诊断生化改变不典型的遗传代谢性肝病,但同一种肝损伤模式也可见于多种疾病 [3] ,如胆汁淤积型病理表现常见于进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis, PFIC)、Alagille综合征(Alagille syndrome, ALGS),贮积型见于糖原累积症(glycogen storage disease, GSD)、溶酶体贮积病,肝细胞脂肪变性见于各型GSD,肝组织炎症见于Wilson病(Wilson disease, WD)等 [4] 。因此,遗传代谢性肝病的诊断常结合临床特征、组织病理学、酶学或基因检测等进行综合性诊断。但不同遗传代谢性肝病的临床表型和肝脏组织学表现可能有重叠,同一疾病在不同患儿中表现可能不同 [5] ,因此本研究对我院行超声引导下经皮肝穿刺活检、临床诊断考虑遗传代谢性肝病且具备组织病理学资料的215例住院患儿的病因进行回顾,总结最终诊断为遗传代谢性肝病的81例患儿临床特点及病理诊断价值,以期帮助临床医师早期识别遗传代谢性肝病和精准掌握肝活检指征。
2. 研究对象与方法
2.1. 研究对象
回顾性研究:以重庆医科大学附属儿童医院2018年1月至2022年6月行超声引导下经皮肝穿刺活检、临床诊断考虑遗传代谢性肝病且具备组织病理学资料的215例患儿为研究对象。本研究经重庆医科大学附属儿童医院伦理委员会审核批准:(2022)年伦审(研)第(331)号。
2.2. 研究方法
记录纳入患儿的性别、年龄、病史、家族史、实验室检查结果、影像学、组织病理学以及疾病最终诊断等情况,探究不同遗传代谢性肝病的临床特点及病理诊断价值。
2.3. 统计学方法
使用SPSS25.0软件进行数据分析,正态分布采用(均数 ± 标准差)表示,偏态分布以中位数表示,计数资料以例数表示,组间比较采用卡方检验,P < 0.05认为差异有统计学意义。
3. 结果
3.1. 临床资料
215例住院患儿中,男性125例,女性90例,男女比1.39:1,年龄0.08~17.50岁,中位年龄5.25岁。综合分析患儿的症状和体征、实验室检查、家族史、组织病理及基因检测等临床资料,215例中有67例最终诊断不明,148例患儿诊断明确。
将诊断明确的疾病分为遗传代谢性肝病和非遗传代谢性肝病,最终诊断非遗传代谢性肝病67例,包括病毒性肝炎48例(如乙型病毒性肝炎35例、EB病毒性肝炎7例、巨细胞病毒性肝炎4例、甲型病毒性肝炎1例、丙型病毒性肝炎1例)、药物性肝损伤16例和肝脏肿瘤性疾病3例;遗传代谢性肝病81例,包括GSD 36例、WD又称肝豆状核变性34例、ALGS 3例、脂质代谢障碍性疾病2例、PFIC 2例、尼曼–匹克病(Ninemann-Pick disease, NPD) 1例、Citrin蛋白缺乏症1例、病因未明IMLD 2例。
最终诊断为遗传代谢性肝病的81例患儿基本资料见表1;特征性肝组织病理表现见图1,电镜表现见图2。

Table 1. The basic data of 81 children with inherited metabolic liver disease
表1. 81例遗传代谢性肝病患儿的基本资料
注:GSD:糖原累积症,WD:肝豆状核变性,ALGS:Alagille综合征,PFIC:进行性家族性肝内胆汁淤积症,IBLD:遗传代谢性肝病,NPD:尼曼–匹克病。

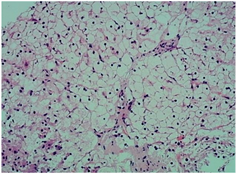 (a) (b)
(a) (b)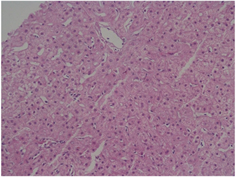
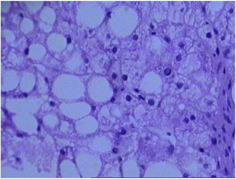 (c) (d)注:(a) 糖原累积症肝组织病理学表现:肝细胞体积明显增大,胞浆淡染,胞界清,呈“植物细胞样”;(b) 肝豆状核变性肝组织病理学表现:肝组织弥漫变性伴局部灶性及点状坏死,偶见空泡状细胞核;(c) 脂质代谢障碍肝组织病理学表现:肝细胞胞浆疏松化,可见大泡性脂肪变;(d) Alagille综合征肝组织病理学表现:部分汇管区未见小胆管。
(c) (d)注:(a) 糖原累积症肝组织病理学表现:肝细胞体积明显增大,胞浆淡染,胞界清,呈“植物细胞样”;(b) 肝豆状核变性肝组织病理学表现:肝组织弥漫变性伴局部灶性及点状坏死,偶见空泡状细胞核;(c) 脂质代谢障碍肝组织病理学表现:肝细胞胞浆疏松化,可见大泡性脂肪变;(d) Alagille综合征肝组织病理学表现:部分汇管区未见小胆管。
Figure 1. Characteristic histopathological changes of liver (HE ×400)
图1. 肝脏组织病理学特征性改变(HE ×400)

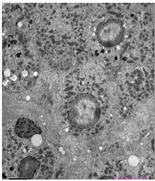
 (a) (b) (c)注:(a) 糖原累积症肝组织电镜:肝细胞胞浆内糖原明显增多,线粒体、内质网等细胞器数量减少;(b) 肝豆状核变性肝组织电镜:多数线粒体大小不等,外形不规则,少部分线粒体内外膜分离,內嵴轻微扩张;(c) 尼曼–匹克病肝组织电镜:肝细胞胞质内含大量空泡结构,空泡内可见髓样小体。
(a) (b) (c)注:(a) 糖原累积症肝组织电镜:肝细胞胞浆内糖原明显增多,线粒体、内质网等细胞器数量减少;(b) 肝豆状核变性肝组织电镜:多数线粒体大小不等,外形不规则,少部分线粒体内外膜分离,內嵴轻微扩张;(c) 尼曼–匹克病肝组织电镜:肝细胞胞质内含大量空泡结构,空泡内可见髓样小体。
Figure 2. Electron microscopic findings of liver tissue
图2. 肝组织电镜表现
36例GSD患儿的首发表现以肝功能异常为主,34例患儿肝脏肿大,13例合并有脾脏肋下可触及,16例有生长发育落后,5例有特殊面容,如圆脸、幼稚面容、泥膏面容及招风耳等。36例患儿均检测肝功能,34例丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(AST)不同程度升高,32例γ-谷氨酰转肽酶(GGT)升高。23例血糖降低,34例患儿检测血氨、乳酸,14例血氨升高,26例乳酸升高。24/36例GSD光镜下表现具有特异性,表现为肝细胞肿胀,胞浆淡染或透亮,胞界清,呈植物细胞样,PAS染色淀粉酶消化前后均阳性,9例光镜表现无特异性,通过超微组织病理诊断。25/29例电镜下表现具有特异性,为肝细胞体积增大,肝细胞胞质内糖原含量明显增多,大片堆积,线粒体等细胞器明显减少且有边聚现象,3例电镜为非特异性表现,通过普通病理诊断。33/36例患儿肝脏组织学具有特异性表现,结合其他临床资料(如转氨酶升高、低血糖、肝脏肿大、乳酸升高、特殊面容、生长发育落后、基因分析等)确诊GSD。
34例WD患儿首发症状以转氨酶升高多见,有4例为双下肢疼痛或浮肿。34例肝功能检查中,26例ALT、29例AST、32例GGT不同程度升高。33/34例患儿检测铜蓝蛋白均降低,其中21例合并24小时尿铜升高,9例可见K-F环。19/34例WD的普通组织病理表现为肝细胞肿胀、脂肪变,偶见空泡状细胞核,汇管区淋巴细胞浸润,偶见豆状核样肝细胞和糖原化核,光镜下无特异性的5例WD电镜有特征性表现。15/22例超微组织病理为线粒体改变(如线粒体肿胀,内嵴扩张,部分线粒体内外膜分离),3例电镜为非特异性肝病的患儿光镜表现具有特征性。24/34例WD患儿有特征性肝脏病理,但10/24例肝脏病理学虽具有特征性,但临床根据其他非组织学资料(如铜蓝蛋白、24小时尿铜、K-F环、头颅MRI表现和基因检测等)已确诊WD,因评估肝脏受累程度或移植前准备而行肝活检。
2例脂质代谢障碍性疾病的患儿入院查体均有肝脏肿大,1例生长发育落后,1例同胞早夭。2例患儿ALT、AST、GGT、血脂、血氨、乳酸均升高,1例串联质谱不排除部分有机酸及脂肪酸代谢异常疾病。2例均未完善电镜,由光镜诊断,肝脏病理表现为肝细胞肿胀,可见大泡性脂肪变,Kupffer细胞增多,肝窦闭塞,汇管区淋巴细胞浸润,纤维组织稍增多。
1例NPD患儿ALT、AST、GGT、血脂、乳酸升高,有生长发育迟缓,骨髓细胞学提示NPD,光镜下表现无特异性,电镜下可见大量空泡结构,空泡内见髓样小体。
3例ALGS均累及肝脏,ALT、AST、GGT不同程度升高,有2例总胆红素升高,以结合胆红素为主,3例血氨、2例乳酸、2例胆汁酸升高,1例患儿光镜表现为汇管区小胆管缺乏,1例电镜检查无特异性,3例患儿基因检查均提示JAG1基因突变,最终通过综合判断临床资料而确诊。
2例PFIC以皮肤、巩膜黄染为首发表现,肝脏肿大,AST、总胆红素(直胆为主)和胆汁酸均有升高,ALT、GGT正常。1例患儿光镜和电镜下为肝细胞淤胆表现,病理诊断意见为需鉴别PFIC,需临床结合病史,建议完善基因检测,本例患儿未完善基因,结合临床其他资料,最终未明确诊断PFIC。另1例患儿未行电镜检查,光镜下表现为肝细胞淤胆,基因检测提示ATP8B1基因纯合变异,结合胆汁淤积的临床表现而确诊PFIC 1型。
2例病因未明IMLD中,1例有肝脾肿大合并苹果脸,1例家族中有早夭。2例ALT、AST升高,1例GGT、总胆红素、血氨、乳酸升高,组织学表现为慢性活动性肝炎和纤维化表现。综合光镜、免疫组化、特殊染色及电镜检查,病理诊断意见均为需鉴别遗传代谢性肝病,结合其他临床资料,其中1例患儿有服用脂溶性维生素10个月,提示药物性肝损伤可能,但结合血尿代筛结果遗传代谢性肝病不能完全除外,基因检测提示致病性未明的变异(FAS、NF1基因),另1例患儿基因检测提示GSD II。型,与临床表型不符,最终2例遗传代谢性肝病均病因不明。
1例Citrin蛋白缺乏症AST、GGT、总胆红素、血氨、乳酸均升高,血串联质谱和尿有机酸检测提示Citrin蛋白缺陷,光镜表现为胆道发育畸形,未完善电镜检查,基因检测发现SLC25A13基因变异,本例患儿通过代谢筛查提供线索,最终通过基因确诊。
3.2. 不同遗传代谢性肝病的主要诊断方式
由于电镜在临床上尚未完全普及,本研究中最终诊断IMLD的81例患儿,并非所有均完善超微组织学检查,各疾病的主要诊断方式并不相同,见表2。在GSD中,光镜电镜结合的诊断率高于光镜,差异有统计学意义(X2 = 16.9, P = 0.003),电镜诊断率高于光镜,差异无统计学意义(P > 0.05);34例WD中,光电镜结合诊断率高于电镜、电镜诊断率高于光镜,但均无统计学差异(P > 0.05)。
Table 2. The main diagnostic methods of inherited metabolic liver diseases (cases)
表2. 各遗传代谢性肝病的主要诊断方式(例)
注:GSD:糖原累积症,WD:肝豆状核变性,ALGS:Alagille综合征,NPD:尼曼–匹克病,PFIC:进行性家族性肝内胆汁淤积症,IBLD:遗传代谢性肝病。
4. 讨论
遗传代谢性肝病往往有临床表型重叠,缺乏特异的实验室检测指标 [6] ,肝穿刺活检有助于指导诊断的方向,本研究215例出院疑诊IMLD的患儿中最终81例诊断为IMLD,说明遗传代谢性肝病临床诊断相对困难,其中组织学诊断IMLD 64例,光镜诊断49例,56例超微组织检查中电镜诊断44例,光镜未诊断的15例在电镜下诊断,电镜未诊断的6例由光镜诊断,故临床上应积极开展电镜检查,光镜结合电镜的肝脏组织学检查可以提高儿童遗传代谢性肝病的诊断率。本研究中3名患儿肝脏病理学提供遗传代谢性肝病的线索,但最终结合临床其他资料及基因检测未明确病因,说明肝穿刺活检有其局限性,部分遗传代谢性肝病的肝脏病理表现为非特异性的组织形态学改变,不能明确诊断,只能缩小诊断范围 [7] ,甚至结合其他检查手段有时也不一定能得到确诊。
GSD主要影响肝脏、肌肉或两者皆有,本研究36例GSD患儿均影响肝脏,多见于婴幼儿和学龄前儿童,发病较早且有相似的表现,如肝脏肿大、转氨酶升高、低血糖、生长发育迟缓、乳酸升高等,其中33例肝脏组织学可诊断,3例组织学未诊断的患儿中有2例未完善电镜检查,可能会降低组织学的诊断价值,其组织学表现具有特异性,光镜下可见肝细胞肿胀,胞浆空淡,核小居中似植物细胞状,电镜下见肝细胞肿大,含有丰富的糖原颗粒,线粒体等细胞器边聚现象,与既往报道一致 [8] 。既往研究报道 [9] 肝脏受累的GSD共同临床特征为低血糖、肝脏肿大、生长迟缓、骨量减少、累及肌肉或心脏等,故临床上身材矮小、生长不良、低血糖、肝肿大伴肝脏转氨酶升高、伴或不伴高脂血症、高乳酸血症等应提高对GSD的怀疑指数,而典型病例根据临床病史、体征及生化检测可作出初步诊断,经肝穿刺活检、基因检测可以确诊,如果严格饮食治疗并定时监测并发症,大多GSD预后良好。
本研究WD患儿的平均发病年龄为8.18岁,多见于年长儿,既往报道儿童发病时的平均年龄为13.2岁 [10] ,其发病年龄和表型取决于个体肝脏对铜毒性的抵抗力和遗传因素 [11] 。34例WD的首发表现以转氨酶升高为主,有相似的异常铜代谢指标,如血清铜蓝蛋白降低和24小时尿铜升高等,但铜蓝蛋白降低并非WD的特异性指标,而临床上年长儿肝功能异常并出现异常铜代谢指标时,仍需警惕WD。本研究中组织学诊断WD 24例,其中10例因评估肝脏受累程度或移植前准备而行肝活检,普通病理结合超微病理的诊断价值最高。WD的肝脏组织学特征多是非特异性的,脂肪变性、炎症、纤维化和肝硬化均可出现,电镜下非特异性的线粒体改变通常被认为是WD相关肝病的典型表现 [12] 。WD的诊断依赖临床症状和体征、实验室检查、影像学、肝脏组织学和基因检测的组合 [10] [13] ,如果怀疑WD,临床病史及非侵入性检查不能做出最终诊断,则需要进行肝活检,多数病理诊断WD需进一步结合临床病史和实验室检查。但是若肝穿刺活检是在未考虑WD时进行,则组织学发现的糖化核和脂肪变性应促使临床医师进一步检测WD相关指标 [14] ,也有一部分肝活检是在确诊WD后需评估病情而进行的。对于大多数WD患者来说,药物治疗或肝脏移植的结果是非常好的,但必须定期进行治疗监测。
本研究中2例脂质代谢障碍和1例NPD患儿均在婴幼儿起病,NPD是脂质代谢障碍的一种,既往报道平均发病年龄1.2 ± 0.6岁 [15] ,总体发病率1/8000~1/5000活产新生儿 [16] [17] 。3例均有肝酶异常和肝脏肿大,其中2例有生长发育迟缓,但生化检查并无特异性,最终由组织学结合临床其他临床资料确诊,组织学多表现为储存型,存储模式包括肿胀和苍白的肝细胞、Kupffer细胞和门静脉巨噬细胞 [3] 。脂质代谢障碍最常见的临床特征是生长发育迟缓、内脏肿大(如肝脏和脾脏肿大) [15] [18] ,肝脏表现从无症状的肝肿大伴轻微的肝酶异常到危及生命的肝功能障碍 [19] ,其病程呈进行性进展,往往导致严重的疾病表现和早期死亡,临床诊断相对困难,其确诊需基于临床表现、家族史,依赖生物标志物、酶活性以及基因分析等,特征性的组织学表现有助于早期诊断,早诊断和早治疗可改善预后。
本研究发现3例ALGS患儿均以胆汁淤积为主要表现,肝活检部分汇管区未见小胆管的1名患儿有前额突出,2名肝脏病例无胆管减少或缺失表现的患儿中1名有先天性心脏病,3例ALGS患儿基因检测均提示JAG1基因突变,JAG1基因突变是ALGS最常见的原因。ALGS常见临床表现为反复发作的胆汁淤积和肝酶异常,肝脏病理特征为小叶间胆管明显减少甚至缺失,胆管缺失更常见于>6个月的儿童,其中<6个月的儿童中只有60%发现胆管缺失,而>6个月的儿童中则为95% [20] ,诊断通过典型临床表现(如慢性胆汁淤积、先天性心脏病、骨骼畸形、角膜后胚胎环、特殊面容等)、生化检测、影像学检查、肝脏病理学改变、家族史和基因突变检测等综合判断,最终预后取决于ALGS累及肝脏和心脏严重程度。
本研究发现2例PFIC患儿在幼儿起病,以黄疸为临床表现,肝脏病理主要为肝细胞淤胆,其中1例基因提示PFIC1型。PFIC的主要临床表现为进行性的黄疸和瘙痒,除了3型GGT升高外,其余各型GGT均正常 [21] 。有研究报道在婴幼儿胆汁淤积症中,PFIC约占9%~13% [22] ,其诊断依赖于患儿的临床表现、实验室检查、影像学和组织学评估等,基因检测可进一步确诊,可以通过肝穿刺活检对其病理特点进行分析,以对家族性肝内胆汁淤积症进行诊断及分型 [23] 。1例Citrin蛋白缺乏症婴儿起病,以黄染为主要表现,肝脾肿大,生化检测无特异性,代谢筛查提示Citrin蛋白缺陷可能,通过基因检测确诊。肝脏脂肪变性是NICCD的一个特征,但本研究中患儿肝脏病理未见脂肪变性,说明即使在胆汁淤积的患儿中未发现肝脏脂肪变性,也不应排除NICCD [24] 。Citrin蛋白缺乏症的诊断主要基于临床病史、血氨基酸、代谢组学、影像和病理学等多项临床资料,基因检测可以确诊 [25] 。
综上,儿童遗传代谢性肝病可在任意年龄起病,转氨酶升高、生长发育迟缓、肝脾肿大、反复发作的胆汁淤积等是常见的临床表现,而肝脏组织学可为遗传代谢性肝病的诊断提供线索,光镜结合电镜的组织学检查可以提高儿童遗传代谢性肝病的检出率,但最终的病理诊断或解释需结合患儿的症状和体征、实验室检查、家族史、影像学以及遗传学分析。
NOTES
*通讯作者。