摘要: 本文采用基于密度泛函理论(DFT)框架下广义梯度近似(GGA)的PBE平面波超软赝势方法,计算了Cu
2CdSnS
4四种结构(W-KS、W-ST、KS和ST)的结构性质、能带结构和光学特性。计算结果表明,Cu
2CdSnS
4的四种结构表现出p型的直接带隙半导体,KS、ST、W-KS和W-ST相的带隙计算值分别为1.1 eV、0.96 eV、1.3 eV和1.0 eV。相比于其他三种结构,KS相结构具有较大的静电常数和吸收系数,有潜力成为薄膜太阳电池的吸收材料。
Abstract:
In this paper, the structural properties, band structures, and optical properties of four structures (W-KS, W-ST, KS, and ST) of Cu2CdSnS4 were calculated using the PBE plane wave ultrasoft pseudopotential method based on the Generalized Gradient Approximation (GGA) under the Density Functional Theory (DFT) framework. The calculation results show that the four structures of Cu2CdSnS4 exhibit p-type direct bandgap semiconductors, with bandgap values of 1.1 eV, 0.96 eV, 1.3 eV, and 1.0 eV for KS, ST, W-KS, and W-ST phases, respectively. Compared to the other three structures, the KS phase structure has a larger electrostatic constant and absorption coefficient, which has the potential to become an absorption material for thin film solar cells.
1. 引言
能源和环境问题是当今人类关注的两大课题,因此近些年来关于新能源、新材料以及环境保护的话题成为讨论的热点。理想太阳电池吸收层材料是带隙在1.1 eV至1.6 eV的直接带隙半导体材料,其中,Cu2ZnSnS4(CZTS)四元半导体材料因其组成元素丰富、高吸收系数和稳定性好而备受关注 [1] 。然而,在制备CZTS薄膜太阳电池时,由于存在带尾、错位缺陷、二次相等问题,使得电池的光电转换效率仅达到12%左右,与理论预测的效率(32.8%)有很大的差距 [2] [3] [4] [5] 。为此,研究人员选取I族、II族或IV族的元素分别取代Cu、Zn或Sn的位置,以消除材料中存在的问题,从而提升CZTS薄膜太阳电池的光电转换效率 [6] [7] [8] 。Ibrahim等人 [6] 制备了Ag或K掺杂的双层CZTS薄膜材料,组装器件的开路电压和短路电流相对于CZTS电池有所改善,实现了8.24%的效率。Zhu等人 [7] 通过混合密度泛函理论计算预测了16种I2-II-IV-VI4 (I = Cu, Ag; II = Sr, Ba; IV = Ge, Sn; VI = S, Se)化合物的能带结构、带隙和光吸收特性。结果发现,Cu2BaGeSe4和Cu2SrSnSe4的带隙值分别为1.60 eV和1.46 eV,有潜力应用于薄膜光伏(单结和多结)领域。Berman等人 [8] 利用第一性原理研究了Na和Ca分别作为Cu和Zn的等价掺杂剂对CZTS缺陷形成、热力学稳定性和电子性能的影响。他们发现,在CZTS内掺杂Na是可行的,而Ca的掺入则是困难的,在CZTS中低钠掺杂是提高CZTS基太阳能电池性能的一个有希望的途径。据目前所知,还未见Cu2CdSnS4不同相结构相关性质对比研究的报道。
本文基于密度泛函理论的第一性原理平面波超软赝势方法先对Cu2CdSnS4的W-KS (Wurtzite-Kesterite,纤维锌矿–锌黄锡矿)、W-ST (Wurtzite-Stannite,纤维锌矿–黄锡矿)、KS(Kesterite,锌黄锡矿)和ST(Stannite,黄锡矿)四种结构进行了优化,然后计算并分析了这四种结构的能带结构和光学性质,筛选出更加适用于太阳电池吸收层材料的构型。
2. 模型与计算方法
本文采用的四种Cu2CdSnS4计算模型如图1所示。整个计算采用了Material Studio软件中的CASTEP软件包进行模拟。CASTEP是一个基于密度泛函理论(DFT)结合平面波赝势方法的从头量子力学计算程序。计算中采用广义梯度近似(GGA)的PBE来处理电子间的交换关联能,电子波函数则通过平面波基矢组展开,并采用超软赝势来描述离子实与价电子之间的相互作用势,选取Cu、Cd、Sn、S各原子价电子组态分别为Cu-3d104s1、Cd-4d105s2、Sn-5s25p2、S-3s23p4。平面波截断能设定为380 eV,布里渊区的K点设定为4 × 4 × 4,自洽收敛精度为5 × 10−7 eV/atom,所使用的晶体结构进行了充分的弛豫。晶体内应力的收敛标准为10−2 Gpa。

Figure 1. Different crystal structures of Cu2CdSnS4: (a) W-ST; (b) W-KS; (c) KS; (d) ST
图1. Cu2CdSnS4的不同晶体结构:(a) W-ST;(b) W-KS;(c) KS;(d) ST
3. 结果与讨论
3.1. 结构性质
不同晶体结构优化后的晶格常数结果如表1所示。从表中可以看出,KS相的晶格常数为:a = b = 0.559 nm;c = 1.118 nm,接近于实验结果 [9] :a = b = 0.553 nm;c = 1.113 nm,这说明所采用的计算方法是可靠的。KS和ST相在垂直方向上数值要大于W-KS和W-ST相,而W-KS和W-ST相在水平方向上数值要大于KS和ST相,且晶胞体积要大一些,其中W-ST相的体积最大,为0.3529 nm3。

Table 1. Lattice constants and volumes of different crystal structures
表1. 不同晶体结构的晶格常数及体积
图2为Cu2CdSnS4的能量–体积(E-V)曲线图。从图中可知,当KS、ST、W-KS和W-ST的能量E分别为−10895.4842 eV、−10895.634 eV、−10895.3659 eV和−10895.3959 eV时,对应的体积V分别为0.3493 nm3、
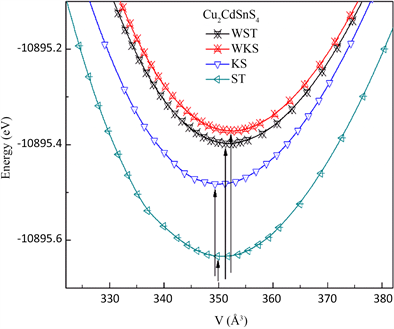
Figure 2. Energy-volume curve of Cu2CdSnS4
图2. Cu2CdSnS4的能量-体积(E-V)曲线图

Figure 3. Band structures of different phases: (a) KS; (b) ST; (c) W-KS; (d) W-ST
图3. 不同相的能带结构:(a) KS;(b) ST;(c) W-KS;(d) W-ST
0.3496 nm3、0.3525 nm3和0.3529 nm3,体系处于稳定状态,其中ST相最稳定。
3.2. 能带结构
不同晶体沿布里渊区高对称点方向的能带结构如图3所示,能量为0的位置为费米能级。四种结构的费米能级位于价带顶,表明导电类型为p型。价带最大值和导带最小值都位于布里渊区的G点,这说明这四种材料属于直接带隙半导体,其计算得到的带隙值小于实验值,这是由于GGA本身存在的缺陷所导致的。通过采用剪刀法进行修正后可得到KS、ST、W-KS和W-ST相的带隙分别为1.1 eV、0.96 eV、1.3 eV和1.0 eV。其中,KS相的带隙计算值与实验报道值1.09 eV非常接近 [9] 。
3.3. 光学性质
复介电函数作为沟通带间跃迁微观物理过程与固体电子结构的桥梁,它反映出固体能带结构及其它各种光谱信息 [10] 。本文中,四种结构的宏观光学响应可以用复介电函数来描述。复介电函数 为实部
和虚部
的和;
,
和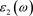 可以用下式来表示:
可以用下式来表示:
(1)
(2)
利用
和
可以求出不同相的吸收系数:
(3)
式中,e、m和M分别表示自由电子的电荷、自由电子的质量和偶极矩阵,i和j表示初态和末态,fi为费米分布,ω表示光子频率,k表示倒易格的向量,
和
表示本征能级。
图4为不同结构的复介电函数随光子能量变化的曲线图。与其他三种结构相比,KS相具有较大的静电常数,这说明KS相这种结构表现出较大屏蔽电荷的能力。随着光子能量的增加,四种结构的实部先

Figure 4. Complex dielectric functions of different structures (Re and Im represent real and imaginary parts, respectively): (a) KS/ST; (b) W-KS/W-ST.
图4. 不同结构的复介电函数(图中Re和Im分别表示实部和虚部):(a) KS/ST;(b) W-KS/W-ST

Figure 5. Absorption coefficients of different structures: (a) KS/ST; (b) W-KS/W-ST
图5. 不同结构的吸收系数:(a) KS/ST;(b) W-KS/W-ST
有微小的增加,随后降低,到达5.7 eV附近时,实部减小到负值以下,这表明这四种结构都具有金属特性,当光子能量大于7.8 eV以后,实部逐渐上升,并接近于1。复介电函数的虚部是表示材料对光子的耗散程度。从虚部的曲线可以看出,这四种结构在光子能量4.6 eV附近出现的较大的峰值,这是由于价带中的电子吸收光子后跃迁至导带所导致。
计算的吸收光谱如图5所示。吸收光谱大致分为3部分:红外吸收区域(<1.6 eV)、可见光吸收区域(1.6~3.2 eV)、紫外光吸收区域(>3.2 eV)。从图4可以看出,四种结构的吸收边与计算的带隙值相对应,在光子能量低于带隙值的区域是透明的,对光基本不吸收,这是因为光子的能量未达到电子所需的激发能,电子未发生跃迁。随着光子能量的逐渐增加,吸收系数也逐渐上升,在光子能量达到7.8 eV附近时,出现最大吸收峰值2.15 × 105 cm−1,这是由于带间发生跃迁所致。在可见光吸收区域,与W-KS、W-ST和ST相的吸收系数相比,KS相具有较大的吸收系数,有潜力成为薄膜太阳电池的吸收层材料。
4. 结论
本文利用基于密度泛函理论的第一性原理平面波超软赝势方法对Cu2CdSnS4的四种结构进行了计算分析,得出了如下结论:
1) 成功优化了Cu2CdSnS4的W-KS、W-ST、KS和ST这四种结构,其中ST相最稳定。
2) Cu2CdSnS4的四种结构表现出p型的直接带隙半导体,KS、ST、W-KS和W-ST相的带隙计算值分别为1.1 eV、0.96 eV、1.3 eV和1.0 eV。
3) Cu2CdSnS4的四种结构都具有金属特性,KS相这种结构具有较大的静电常数,表现出较大屏蔽电荷的能力。
4) 在紫外光区域,Cu2CdSnS4的四种结构由于带间发生跃迁,使得在7.8 eV附近出现最大吸收峰值2.15 × 105 cm−1;在可见光区域,与W-KS、W-ST和ST相的吸收系数相比,KS相具有较大的吸收系数。
基金项目
本工作得到云南开放大学校级科研基金项目(No. 21YNOU17)的赞助。