1. 引言
全基因组关联分析(Genome-wide Association study, GWAS) [1] [2] [3] 是疾病关联分析中应用较为广泛的分析方法。GWAS主要是研究单核苷酸多态性(Single-nucleotide polymorphisms, SNP)与疾病的关联分析。研究人员已经利用GWAS识别出很多与疾病或复杂性状相关的SNP。但大多数SNP的效应可能很小,也不能完全解释疾病或复杂性状的遗传效应。
进一步的研究表明基因与环境因素的交互效应对复杂性状或疾病的解释也起着极为重要的作用。由于基因主效应对于复杂性状或疾病的解释比较有限,基因与基因的交互效应(gene-gene interaction, GGI)分析得到了越来越多的研究 [4] [5] ,基因之间的相互作用能够影响复杂性状的表现,包括一些常见疾病。理解这些交互效应对于疾病的发病机制和个体之间的差异至关重要。基因交互效应有助于揭示遗传学中的复杂性,帮助我们更好地理解基因对生物体特征和疾病易感性的贡献 [6] 。
多数疾病都是由多个基因和环境因素相互作用引起的。基因之间的复杂交互可以导致疾病的多样性和不同个体之间的差异。特定基因的单一变异可能并不足以显著增加疾病风险,但是当这些基因与其他基因相互作用时,可以导致更高的疾病风险。同时基因交互作用可以从网络生物学的角度来理解 [7] ,即基因以复杂的网络方式相互连接。这种网络的破坏或改变可能与疾病的发生和发展密切相关。不同个体之间的基因组组合各异,这意味着对于相同疾病,可能存在不同的遗传机制。总体而言,深入研究基因与基因之间的交互作用有助于揭示疾病的遗传基础,为个性化医疗和疾病治疗提供更深入的理解和方法。
在研究遗传变量之间的交互效应中,基因与基因交互效应的分析涉及多种方法,研究人员提出了几种方法 [8] [9] 来检测SNP-SNP的交互效应,通过这种关联分析,研究人员可以检测基因变异与疾病之间的关系。这可以通过单一基因关联(单基因研究)或考虑多个基因同时的关联来进行(多基因关联研究) [10] :如基于熵的统计、Logit模型等 [11] [12] [13] ;其他技术同时也包括多因素降维(Multifactor dimensionality reduction, MDR)、BOOST、RRIntCC、GenEpi [14] [15] [16] [17] 以及一些加速方法 [18] 。这些方法有几个潜在的优点。首先,它通常含有更少的基因,可减少成对检验的数量。由于交互效应的存在,这些方法也适用于基因的主效应研究。此外,生物学的一些先验(例如,关于蛋白质与蛋白质交互效应(Protein-Protein Interaction Network, PPI)或已知的基因关联信息)也可以很容易地引入到研究中。同时使用机器学习算法,如随机森林、支持向量机、神经网络等,也可以来探索基因之间的复杂交互效应。不过,这些方法也存在一些挑战和不足:基因交互效应分析往往涉及大量基因组数据,导致维度灾难 [19] [20] ,使得分析变得更为复杂。在分析多个基因或变异时,需要考虑多重比较问题,以避免虚假的关联结果。机器学习等复杂模型的解释性可能较差,难以理解具体的基因之间交互的生物学意义。最近,研究人员提出了一种称为AGGrGATOR [21] 的方法,该方法在标记水平上结合P值检验基因交互效应的显著性,这是用于检测定量表型下的交互效应的策略 [22] 。也有研究人员提出了一种基于熵的非参数方法GBIGM [23] 检测交互效应。在生物学中,植物的性状 [24] 也受到许多基因与基因之间交互效应的影响。有研究表明EYA4基因和GRHL2基因之间的交互效应与噪声性耳聋(Noise-induced hearing loss, NIHL)的发生存在关联 [25] 。基因之间的交互效应得到了越来越多的研究验证。这些方法遇到的主要问题之一是多重检验的校正会导致的高阶效应或成对检验的显著性受到影响。
同时近年来,越来越多的高频观测数据以函数曲线的形式出现,比如每分钟股票价格数据、汇率数据、气温数据、小时PM2.5数据、光谱数据等,这些数据常用的分析方法是函数型数据分析(Functional Data Analysis, FDA) [26] 。大数据时代传统的结构化数据也从简单的点数据,扩展到区间数据、符号数据和函数型数据等 [27] 。函数型数据分析已经成为统计分析中越来越重要的研究方向。函数回归分析也得到了越来越广泛的研究和应用。
从很多已有的研究结果中可以看到,与疾病相关联的基因数目一般都比较大,若进一步考虑交互效应将使得模型待估参数的个数变得非常高,这给统计分析带来了很大的挑战。另外有些主效应和交互效应比较弱,检测比较困难。综合来看,要解决该问题需要综合运用多种方法来克服挑战,如何降维以及如何处理交互效应,在这里本文利用函数回归模型对主效应和交互效应进行整体考虑,基因组数据通常是高维度的,处理这些大规模的遗传信息需要发展更有效的统计和计算方法。模型中的函数效应可通过常用的基展开进行估计,这也可以有效地降低的降低模型待估参数的维数,也更有利于检测出弱效应。本文将基因位点(loci)连续化 [28] ,把基因的主效应和基因与基因的交互效应进行函数化处理,这种扩展的Cox模型对于生存分析中考虑基因与基因之间的交互作用是一种常见的方法。当考虑基因与基因的交互作用时,Cox比例风险模型可以包含这些交互项。交互作用项评估基因之间的相互作用对生存时间的影响是否显著。基于此,我们建立了函数型交互效应Cox比例风险模型。
2. 模型介绍
2.1. Cox比例风险模型
在这里我们知道Cox比例风险模型的基本形式为:
在上式中,
表示自变量,
为自变量的偏回归系数。
表示t时刻的基准风险率函数。
2.2. 函数交互效应Cox比例风险模型
假设有m个SNP,对应的基因位点为
,在具体分析中可以将基因位点进行归一化处理。
令
表示个体i的生存时间,
表示其右截尾时间。设
是观察到的时间,
。此外,设
表示m个SNP的基因型,其中
是
位置的次等位基因(Minor Allele)的数目,并且
表示协变量向量。
对于上面的数据,常规的Cox比例风险模型为
当m较小时,上面的模型可以用于交互效应和主效应分析;当m很大时,则该模型可能会因为参数过多而无法估计,即使能够估计模型参数,其功效也会较低。
为了有效降低模型参数个数和提高功效,基于Zhang et al. (2021) [28] ,本文提出如下函数型Cox比例风险模型:
(1)
其中
是基线风险函数,
是关于u的主效应函数,
,
是关于u、v的交互效应函数。
在模型(1)中,假设主效应函数
是光滑的,即
是关于u的连续函数。它可以用B样条函数展开。假设
由一系列
基函数
展开为
,其中
是系数的
向量,
。在这里我们利用B样条基:
来展开主效应函数。
设
是一系列K基函数,如B样条基函数。设
表示含有
的
矩阵,
。利用已有结果 [24] [25] [26] ,
可由下式进行估计:
(2)
对于交互作用,根据对称性,我们有
对于交互效应函数,我们利用张量积 [29] 对
进行展开。
张量积使一维P样条自然地延伸到二维,假设除了x之外,还有第二个解释变量v。对于
,有数据三元组
。寻找一个光滑的曲面
,它可以给出数据一个很好的近似。设B,
是沿x的B样条基,B,
是沿v的B样条基。构成KL张量积
。设
是系数的
矩阵。然后,对于给定的A,我们得到
的拟合值是:
因此在这里我们设
是系数的
矩阵,则
。令
(3)
模型(1)可利用上面的基展开表示为
(4)
其中
,
3. 模拟研究
3.1. 模拟设置
在下面的模拟中,显著性水平为0.05、0.01和0.001;考虑三种删失情况:(a)
,(b)
,(c)
。基因型是从6 kb和9 kb亚区的SNP中选择的,在下面模拟中的模型含有100个SNP。样本量为
和
,每种设置组合下,重复进行4000次模拟。
模型中的SNP主效应
,其中
(Minor Allele Frequency,次等位基因频率)是第j个SNP的次等位基因频率。罕见变异(Rare Variant)通常是指MAF ≤ 0.05的SNP。在模拟中,考虑10%的变异在6 kb和9 kb区域被选择作为关联变异,对于关联变异考虑两种组合:1、10%的常见变异(common variant)和90%的罕见变异;2、100%的罕见变异。对于6 kb亚区,
;对于9 kb来说,
。常数k和遗传效应大小随着区域大小的增加而减小。对于第一种变异组合,在6 kb亚区,k = 5.5;在9 kb亚区,k = 6;对于所有SNP为罕见变异的情况,在6 kb亚区,k = 1.25;在9 kb亚区,k = 1.5。
3.2. 第I类错误率
生存时间由如下模型生成:
其中
是均匀分布的随机变量U(0,1),
是从正态分布
到模型的连续协变量。基于所设置的模型和模拟参数,对每个产生的数据利用提出的交互效应模型(3)进行分析,并利用LRT检验累积交互效应是否显著。
在附表1中(详见附录),所有变异(常见和罕见)都用于在零假设下生成基因和生存时间数据。显著性水平α分别取值为0.05,0.01,0.001。总的来说,所提出的模型能够较好的控制了第I类错误率。这两个模型在6 kb和9 kb的区域尺寸以及2000和2500的样本量下都是稳定的,结果非常相似。模拟结果表明所提出的模型在区域大小、检查方案、名义水平均是稳定的和有效的。
3.3. 功效模拟分析
生存时间由如下模型产生:
在这里
,
对于6 kb亚区,
;对于9 kb来说,
。当罕见变异占比为90%,常见变异占比为10%时,在6 kb亚区,
;在9 kb亚区,
;对于所有SNP为罕见变异的情况,在6 kb亚区,
;在9 kb亚区,
。
当样本大小为2000时,功率如图A、B所示,6 kb和9 kb分别代表不同的区域,当样本大小为2500
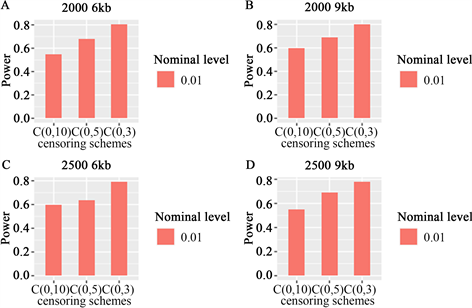
Figure 1. When the sample size was 2000 and 2500, and the region size was 6 KB and 9 kb, the LRT statistic was a potential function at α = 0.01, where some variants were common and others were rare. The Order of B-spline basis is 4
图1. 当样本量为2000和2500,区域大小为6 kb和9 kb时,LRT统计量在α = 0.01时的势函数,其中一些变异是常见的,其余变异是罕见的。B样条基的阶为4

Figure 2. When the sample size was 2000 and 2500, and the region size was 6 KB and 9 kb, the LRT statistic was a potential function at α = 0.01, and all variants were rare
图2. 当样本量为2000和2500,区域大小为6 kb和9 kb时,LRT统计量在α = 0.01时的势函数,所有变异都是罕见的
时,功率如图C、D所示,6 kb和9 kb分别代表不同的区域;在图A-D中,常见变异和罕见变异占比分别为90%和10%。在图E-H中,所有SNP为罕见变异。
图1四个图的每个图中,比较了三种不同删失率、不同样本容量、不同区域的功效,所提出的Cox比例风险模型均能比较有效的检测出累积交互效应,功效随着样本容量的增加及删失率的减少而增加。图2中分析了变异全部为罕见变异的情况,从图中可以看到与图1类似的功效表现。函数交互效应Cox比例风险模型LRT统计量是非常稳定的,因为它们不强烈依赖于基因型数据是否平滑。
4. 案例分析
在AREDS (Age-Related Eye Disease Study年龄相关眼病研究)中的应用
利用所提出的函数交互效应Cox比例风险模型来分析AREDS [24] 数据(年龄相关眼病研究组,1999)。AREDS是一项临床试验,旨在了解影响老年黄斑变性和白内障的风险因素,这两种疾病是导致老年人视力丧失的主要原因。共有2911人被纳入这项分析,其中1650人为男性,1261人为女性。2911人的平均年龄为68.65岁,标准差为4.92岁。本文仅研究左眼的视力数据,左眼的删失率为76%。在分析中,我们将年龄和性别作为协变量进行调整,且每个个体都有长期的表型数据。根据Seddon等人在2007的研究中表明在AMD (age-related macular degeneration老年黄斑变性)疾病研究中有两个基因区域,CFH和ARMS2,与AMD的风险及其进展相关。这里只研究CFH基因区的交互效应。在CFH基因区域有103个罕见变异,59个常见变异,CFH基因的常见和罕见变异都会影响AMD的进展。在这里我们在样条函数的处理中除了使用B样条之外还使用了傅立叶样条,此外我们还利用Logistic模型 [30] 对交互作用的显著性进行分析,结果见表1,显示这个基因区内的SNP之间的交互作用对老年黄斑变性风险有显著影响,对于结果来说,我们提出的带有B样条的Cox模型效果更为显著,Logistic模型的显著性明显不如我们所提出的模型。

Table 1. Interaction within CFH gene fragments
表1. CFH基因片段内的交互作用
注:罕见变异定义为MAF ≤ 0.05的变异,常见变异定义为MAF > 0.05的变异。CFH × CFH表示CHF基因片段的SNP之间的交互效应。
5. 总结
本文在Bingsong Zhang等研究基础上对基因–基因交互效应的检测上进行了拓展。与疾病相关联的SNP数量比较大时,它们之间的交互效应维数将会极大的增加,有些交互效应也比较微弱,这使得交互效应的分析变得较为困难。本文在基因主效应函数化的基础上把基因–基因交互效应也进行函数化,通过基展开,可有效降低待估参数的维数,也可对交互效应进行有效检测。在前面模拟中,SNP的个数为100,两两交互效应的数量高达4950,待估参数的个数远大于观测值的数量。通过将主效应和交互效应进行函数化,极大地降低了待估参数的个数。从模拟结果可以看出第I类错误率在各种情形下都得到了很好的控制。此外,在功效的模拟研究分析中,所提出方法可有效地检测出基因–基因交互效应。第四部分将所提出的函数型Cox模型应用在了眼部疾病的数据中,分析结果表明我们的模型验证了CFH基因会影响老年黄斑变性的进展,其基因片段内的SNP之间的交互效应也会影响老年黄斑变性的进展,不论它是常见变异还是罕见变异。这显示了加入交互效应的模型的有效性。
本文的研究对于基因–基因交互效应的分析主要检验的是累积交互效应,但是部分交互效应可能会不显著,也不应该放入模型进行分析,否则会降低检验的效率。另外,高阶交互效应、基因–环境因子等还没有进行考虑,这些均可作为未来研究的内容。
基金项目
本文由中央高校基本科研业务经费(SWJTU, 2682021ZTPY078);国家自然科学基金面上项目(51578471)资助。
附录
Appendix Table 1. Type I error rates for two sample sizes
附表1. 两种样本容量下的第I类错误率
(当区域大小为6和9 kb时,名义水平α = 0.05,0.01,0.001,0.0001的Cox FR LRT统计量的第I类错误率,情景1表示有一些变体是常见的,其余的是罕见的,情景二表示所有变异都是罕见的。)