1. 引言
多环芳烃(Polycyclic aromatic hydrocarbons, PAHs)是由两个或两个以上苯环稠合而成的一类芳香族化合物,通常为有机物在高温缺氧状态中裂解产生的活跃自由基及碎片微粒键合而成 [1] 。目前已知2-7环结构的多环芳烃就有几百种,某些结构的多环芳烃具有致突变性和致癌性 [2] [3] [4] 。煤和石油等有机高分子化合物在不完全燃烧时易产生多环芳烃,并广泛分布于自然界中,在一定条件下进入大气、水源、土壤等环境介质后很难降解,通过人体吸入或食物链作用,在生物体内累积 [5] [6] 。当其进入身体,将直接影响接触人的身体健康,甚至会导致癌症等恶性疾病的发生,因此研究多环芳烃的测定具有非常重要的意义 [7] 。
美国环保署已将萘、苊、二氢苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、䓛、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-cd]芘、二苯并[a,h]蒽、苯并[ghi]苝16种PAHs确定为优先控制的环境污染物 [8] 。2001年原国家环保总局在《国家环境科技发展“十五”计划纲要》就指出多环芳烃等有毒有害化学品污染已对环境和人体健康构成明显威胁 [9] 。目前,对水中16种多环芳烃的检测方法越来越多,仅高效液相色谱法而言,2006年发布的《生活饮用水标准检验方法》(GB/T 5750.8-2006) [10] 对水中多环芳烃的分析就采用高效液相色谱法,2009年环境保护部发布《水质多环芳烃的测定液液萃取和固相萃取高效液相色谱法》(HJ 478-2009) [11] (以下简称环境标准)对水中16种多环芳烃检测作了明确的规定。已报道的文献对高效液相色谱法测定水中16种多环芳烃也有一些,随着技术条件的成熟,2023年国家又重新修订发布《生活饮用水标准检验方法》(GB/T 5750.8-2023) (以下简称国家标准) [12] 。对于国家标准和环境标准方法,部分检测机构不能正确理解和应用。在国家标准方法实施之前,为了更好区别国家标准和环境标准的不同,解决水中多环芳烃检测过程的疑惑,本文对高效液相色谱测定水中16种多环芳烃的国家标准和环境标准在适用范围、样品采集与保存、样品前处理、色谱条件、检出限、精密度和准确度等内容进行了比对分析,并采集实际样品进行测试比对。探讨两种方法的优缺点及方法应用过程的建议,为检测实验室在选择方法和样品分析过程中提供参考。
2. 参数比对
国家标准测定的多环芳烃:萘、苊烯、苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、䓛、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-cd]芘、二苯并[a,h]蒽、苯并[ghi]苝;环境标准测定的多环芳烃:萘、苊、二氢苊、芴、菲、蒽、荧蒽、芘、苯并[a]蒽、䓛、苯并[b]荧蒽、苯并[k]荧蒽、苯并[a]芘、茚并[1,2,3-cd]芘、二苯并[a,h]蒽、苯并[ghi]苝;从测定的项目可以看出,国家标准和环境标准测定项目均为16种多环芳烃,有的名称不一样,经核实苊烯别名为二氢苊,因此两种方法所规定16种多环芳烃项目一样。两种方法在适用范围、样品采集、样品前处理、色谱条件、检出限、精密度和准确度有所不同。
2.1. 适用范围
国家标准规定了用于生活饮用水及其水源水;环境标准适用于饮用水、地下水、地表水、海水、工业废水及生活污水,其中液液萃取适用于饮用水、地下水、地表水、工业废水及生活污水,固相萃取适用于清洁水样。从适用范围来看,环境标准适用范围更广泛。
2.2. 样品采集和预处理
2.2.1. 国家标准的水样采集和保存
采集水样时,若含有余氯,先加抗坏血酸于采样瓶中(每升水样加0.1 g抗坏血酸,余氯含量高时可增加用量)。采1~4 L水样,加磷酸调节pH < 2,密封;水样于0℃~4℃避光保存,保存时间7天。
2.2.2. 环境标准的样品采集与保存
样品必须采集在预先洗净烘干的1 L或2 L具磨口塞的棕色玻璃细口瓶。采样前不能用水样预洗采样瓶,以防止样品的沾染或吸附。采样瓶要完全注满,不留气泡。若水中有残余氯存在,要在每升水中加入80 mg硫代硫酸钠除氯。样品采集后应避光于4℃以下冷藏,在7 d内萃取,萃取后的样品应避光于4℃以下冷藏,在40 d内分析完毕。
2.2.3. 国家标准的样品预处理
取500 mL水样于广口玻璃瓶或聚四氟乙烯的瓶中,加入10 mL甲醇,摇匀。依次向苯乙烯二苯乙烯聚合物柱(填料250 mg,容量6 mL)加入10 mL二氯甲烷、6 mL甲醇、6 mL水活化。以3~6 mL/min速度上样,为降低瓶壁对目标物的吸附,上样结束后用10 mL 50%甲醇水溶液(pH < 2)润洗样品瓶,继续上样;用6 mL 80%甲醇水淋洗(流速 ≤ 3 mL/min),淋洗结束后用洗耳球按压挤干固相萃取柱上液体(不宜负压抽干,否则会造成萘等部分目标物回收率偏低);用10 mL二氯甲烷洗脱(流速 ≤ 1 mL/min)或分次浸泡洗脱(5次 × 2 mL,浸泡2 min),洗脱液用10 mL玻璃试管收集;向洗脱液表面滴加100 μL吐温−20的甲醇溶液后氮吹,小流量氮吹至近干,用50%乙腈水溶液1.0 mL复溶,在旋涡振荡仪震荡混匀,将浓缩的洗脱液通过滤膜(尼龙滤膜或亲水性滤膜)过滤后转移至进样瓶中,进样分析。
2.2.4. 环境标准的液液萃取
量取1000 mL水样,倒入2000 mL的分液漏斗中,加入50 μL十氟联苯,加入30 g氯化钠,再加入50 mL二氯甲烷 ,振摇5 min,静置分层,收集有机相,放入250 mL接收瓶中,重复萃取两遍,合并有机相,加入无水硫酸钠至有流动的无水硫酸钠存在。放置30 min,脱水干燥。用浓缩装置浓缩至1 mL,待净化。如萃取液为二氯甲烷,浓缩至1 mL,加入适量正己烷至5 mL,重复此浓缩过程3次,最后浓缩至1 mL,待净化。饮用水和地下水的萃取液可不经过柱净化,转换溶剂至0.5 mL直接进行HPLC分析。地表水和其他萃取液的净化:用1 g硅胶柱作为净化柱,将其固定在液液萃取净化装置上。先用4 mL淋洗液冲洗净化柱,再用10 mL正己烷平衡净化柱(当2 mL正己烷流过净化柱后,关闭活塞,使正己烷在柱中停留5 min)。将浓缩后的样品溶液加到柱上,再用约3 mL正己烷分3次洗涤装样品的容器,将洗涤液一并加到柱上,弃去流出的溶剂。被测定的样品吸附于柱上,用10 mL二氯甲烷/正己烷(1 + 1)洗涤吸附有样品的净化柱,收集洗脱液于浓缩瓶中(当2 mL洗脱液流过净化柱后关闭活塞,让洗脱液在柱中停留5 min)。浓缩至0.5~1.0 mL,加入3 mL乙腈,再浓缩至0.5 mL以下,最后准确定容到0.5 mL待测。
2.2.5. 环境标准的固相萃取
将固相萃取C18柱安装在自动固相萃取仪上或手动固相萃取装置。先用10 mL二氯甲烷预洗C18柱,使溶剂流净。接着用10 mL甲醇分两次活化C18柱,再用10 mL水分两次活化C18柱,在活化过程中,不要让柱子流干。在1000 mL水样(富集所用水样体积根据水质情况可适当增减)中加入5 g氯化钠和10 mL甲醇,加入50 μL十氟联苯,混合均匀后以5 mL/min的流速流过已活化好的C18柱。用10 mL水冲洗C18柱后,真空抽滤10 min或用高纯氮气吹C18柱10 min,使柱干燥。用5 mL二氯甲烷洗脱浸泡C18柱,停留5 min后,再用5 mL二氯甲烷以2 mL/min的速度洗脱C18柱,收集洗脱液。先用10 mL二氯甲烷预洗干燥柱,加入洗脱液后,再加2 mL二氯甲烷洗柱,用浓缩瓶收集流出液。浓缩至0.5~1.0 mL,加入3 mL乙腈,再浓缩至0.5 mL以下,最后准确定容到0.5 mL待测。
可以看出,两种方法的样品都要求低温避光保存,对预处理过程规定得很细致;国家标准预处理方式只有固相萃取,但用水量较少;环境标准有液液萃取和固相萃取,需加入十氟联苯作为替代物,液液萃取适用于各种水样,固相萃取只适用于清洁水样,但取样量较大。
2.3. 色谱条件
表1为两种方法所推荐色谱条件和参数,表2为流动相洗脱程序。从表1得,国家标准推荐使用PAH-C18色谱柱为多环芳烃专用柱,环境标准推荐的色谱柱填料为ODS属于C18反向柱,但规格和型号一致;从表2知,在洗脱程序方面国家标准只推荐1种,环境标准推荐2种,但从洗脱时间来看,国家标准5分钟后就开始进行梯度洗脱,使用时间更短。
2.4. 检测器
国家标准指出苊烯在荧光检测器没有响应,要求用紫外检测器和荧光检测器进行串联检测;环境标准里指出苊在荧光检测器没有响应与国家标准规定有出入,经使用单标准溶液进行实验验证及查阅土壤和沉积物多环芳烃的测定高效液相色谱法(HJ 784-2016) [13] 等,确定苊在荧光检测器有响应,苊烯在荧光检测器没有响应,因此苊烯需要紫外检测器进行检测,环境标准推荐紫外检测器对16种多环芳烃进行检测,使用荧光检测器对15种多环芳烃(除苊烯外)进行检测。
2.5. 检出限
根据方法提供的检出限见表3。从表3可以看出,国家标准只规定了1种检出限,环境标准规定了4种检出限,环境标准固相萃取检出限较低。
2.6. 精密度和正确度
加标回收率范围和相对标准偏差是根据标准方法中不同实验室测定结果进行计算,数据见表4。从表4知,国家标准的加标回收更稳定,精密度更高。
2.7. 质量控制和质量保证
国家标准在质量控制和质量保证未单独提出明确规定,采用的是统一的质量控制标准;环境标准在空白试验和加标回收作出了明确规定,空白试验均要低于方法检出限,空白加标各组分的回收率在 60%~120%,替代物十氟联苯回收率在50%~130%。
3. 实验比对
为了更好提供直观的参考,本文按照两种方法的要求对实样进行预处理后,在安捷伦1260Ⅱ液相色谱仪分析测定。在色谱柱方面选择用了C18反向柱和PAH专用柱进行对比,PAH专用柱比C18反向柱分离度更好。在梯度洗脱方面国家标准方法需延长洗脱时间目标物质才能全部洗脱出来,环境标准中的甲醇洗脱方式用时过长不建议使用,采用乙腈和水洗脱的方式虽部分目标物质受负峰影响但只需进行微调即可。
3.1. 色谱图
国家标准要求使用紫外和荧光检测器串联使用,紫外检测器主要用来测定苊烯(二苊氢),荧光检测器测定其他15种多环芳烃。图1为紫外波长228 nm处谱图,图2为荧光谱图。从图可以看出紫外谱图分离度好,杂峰少,由于使用低浓度,响应值低;但荧光谱图响应值高,有杂峰,各组分保留时间稳定,不影响定性,适合低浓度饮用水检测。
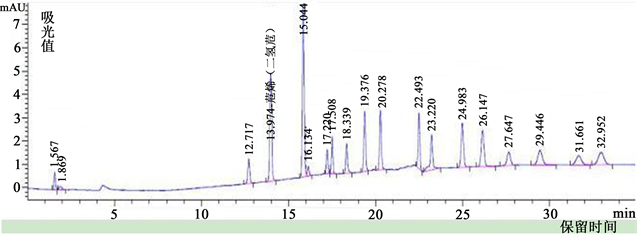
Figure 1. Acenaphthylene (Diacenaphthyl Hydrogen) 0.10 µg/mL standard solution spectrum at UV wavelength 228 nm
图1. 苊烯(二苊氢) 0.10 µg/mL标准溶液紫外波长228 nm处谱图
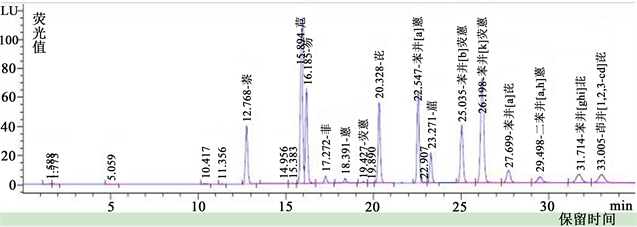
Figure 2. Fluorescence spectrum of 0.10 µg/mL standard solution of 15 PAHs
图2. 15种多环芳烃0.10 µg/mL标准溶液荧光谱图
图3为环境标准中16种多环芳烃1.0 µg/mL和替代物十氟联苯2.0 mg/L标准溶液紫外波长220 nm处谱图,图4为1 L水样分别加入16种多环芳烃标准溶液1.0 µg后,浓缩至0.5 mL的色谱图。从图3和图4可以看出,16种多环芳烃分离度好,响应值高,实际样品有一定杂峰,但各组份保留时间也稳定,定性较好,能满足检测要求。
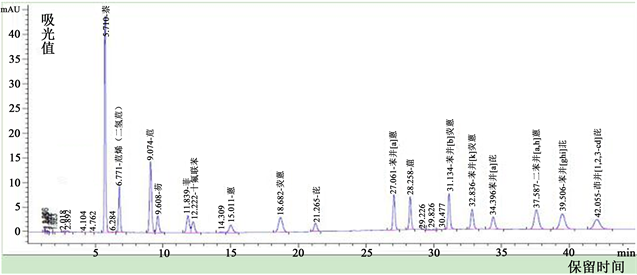
Figure 3. 16 kinds of polycyclic aromatic hydrocarbons 1.0 μg/mL and decafluorobiphenyl 2.0 mg/L standard solution UV spectrum at 220 nm
图3. 16种多环芳烃1.0 µg/mL和十氟联苯2.0 mg/L标准溶液紫外波长220 nm处谱图
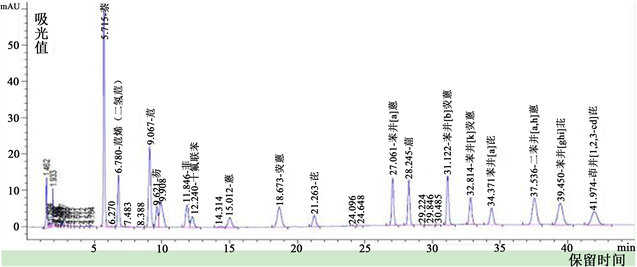
Figure 4. The UV chromatograms of 16 actual PAH samples spiked at 1.0 µg/L and concentrated to 0.5 mL
图4. 16种多环芳烃实际样品加标1.0 μg/L浓缩至0.5 mL的紫外色谱图
3.2. 校准曲线
用16种多环芳烃200 μg/mL的标准溶液(LGC, DRE-A50000533AL)按照国家标准方法要求准配制成0.000、0.020、0.050、0.100、0.150、0.200 μg/mL的标准系列,按照环境标准方法要求配制成0.10、0.50、1.00、5.00、10.0μg/mL的标准曲线系列,分别进入预设定好条件的液相色谱仪进行测定,结果见表5。从表5得,两种方法各组份线性良好,国家标准使用低浓度系列,符合饮用水检测实际;而环境标准线性范围宽,总体线性优于国家标准,适用于各种浓度的水样分析。

Table 5. 16 kinds of polycyclic aromatic hydrocarbons calibration curve record table
表5. 16种多环芳烃校准曲线记录表
3.3. 实际水样
采集实际样品分别平行测定6次,结果见表6。从表6得,两种方法均有很好的回收率,国家标准菲回收率低,环境标准萘回收率低。
Table 6. Actual water sample measurement results table
表6. 实际水样测定结果表
4. 结论
从各项参数和实样测定结果来看,两种方法均有很强适用性和针对性。国家标准方法(GB/T 5750.8-2023)在测定水中16种多环芳烃中具有样品使用量少,分析速度快,操作简单,试剂和标准物质用量少,加标回收稳定,精密度高等优点,但仅限于饮用水及其水源水。环境标准方法(HJ 478-2009)具有适用范围广,线性范围宽,预处理方法多,可选多种检测方式,固相萃取检出限较低,可根据不同水样灵活应用,但样品用量大,试剂和标准物质用量多,固相萃取的奈和替代物十氟联苯回收率不理想。
5. 建议
建议检测实验室要根据开展工作的实际情况对检测方法进行认证,如果是单纯检测饮用水中的多环芳烃只需认证国家标准即可,而社会检测机构面向市场多元化时,尽可能两种方法都进行认证,根据样品的种类来选择不同的分析方法。
参考文献
NOTES
*第一作者,陆文灵,男,工程师,主要从事生态环境监测有机物分析工作。
#通讯作者,邵菠昌,女,工程师,主要从事生态环境监测工作。