1. 引言
木质素是生物质重要组成部分,其含量丰富,是目前可再生芳香族化合物的最大来源 [1] [2] 。木质素是一种复杂、无定形、三维结构的高分子化合物 [3] [4] ,由于其化学结构复杂导致难以解聚,被认为是低价值的原料 [5] [6] [7] [8] 。最常见的木质素利用方式有热降解法 [9] 、快速热裂解、生物降解法、氧化降解法等 [10] [11] 。相较于这些木质素利用技术,电化学氧化技术具备成本低、无污染、反应条件温和、稳定可控等优势。电化学氧化降解木质素耦合电解水一方面可以通过阳极电氧化降解木质素副产高值化学品,另一方面可以加快阳极反应、降低阳极过电位和整体电解能耗、提升产氢效率 [12] [13] 。目前,国内外学者利用各种方法对木质素的降解机理进行了模拟计算 [14] [15] ,蒋晓燕 [16] 对α,β-双醚型木质素三聚体进行了研究,Cβ-O和Cα-Cβ均裂是竞争反应,而Cα-O均裂在初步裂解反应中是最主要的反应,Younker等认为β-5型木质素二聚体中Cα-O键和Cα-Cβ键裂解是主要途径,而且不同取代基对两种键的键解离能会产生影响。田红等 [17] 对5-5’型木质素二聚体进行研究,发现其苯环3号位上的甲氧基具有很强的活性,O-CH3键以及Caromatic-OCH3键较易断裂。相较于多聚体模型化合物和单体模型化合物,采用木质素二聚体模型化合物进行研究的较多,二聚体木质素模型化合物不仅比单体更能清晰地反映木质素中苯环之间的连接键,而且比多聚体结构简单,更加易于合成和获得。
在此基础上,本文选取由愈创木基丙烷结构单体聚合而成的愈创木基木质素(Guaiacyl lignin,G-木质素)二聚体作为研究对象,从分子层面深入了解木质素电化学氧化过程以及产物形成的机理,构建木质素理论模型,采用杂化密度泛函B3LYP/6-31G(d)基组对初始构象进行优化和计算,通过计算键解离能、HOMO和LUMO轨道活性点位分析和势能分析等方法研究木质素二聚体模化物反应路径,为木质素电化学氧化耦合电解水制氢的优化提供理论支撑。
2. 实验部分
2.1. 木质素二聚体模型化合物构建
在木质素模型化合物的研究中,国内外学者采用二聚体木质素模型物进行研究的非常多。木质素二聚体是木质素内具有典型连接键且结构较为简单的模型化合物,由愈创木基、紫丁香基和对羟基苯基三种基本结构单元,通过醚键、β-O-4或C-C键把两个单体结构连接而成 [18] ,如图1所示。
 对羟苯基丙烷愈创木基丙烷紫丁香基丙烷
对羟苯基丙烷愈创木基丙烷紫丁香基丙烷
Figure 1. The three basic structural units of lignin
图1. 木质素的3种基本结构单元
其中C-C键是主要的连接方式。目前,在木质素模型化合物中研究中,常用二聚体模化物来进行研究。同单体相比,二聚体模化物更能真实的反映出木质素内部键型的连接方式,且相对于多聚体模型化合物,二聚体模化物裂解的产物种类更少,较容易得到准确的理论分析,从而能够深入了解木质素的电化学氧化过程。本文选择由愈创木基丙烷结构单体聚合而成的愈创木基木质素(Guaiacyl lignin,G-木质素)二聚体为研究对象,构建愈创木基丙烷木质素二聚体模型化合物(图2)。
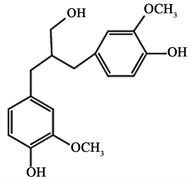
Figure 2. Guaiac-based propane lignin dimer model compound
图2. 愈创木基丙烷木质素二聚体模型化合物
2.2. 密度泛函理论以及计算方法
密度泛函理论是物理、化学量子理论计算使用最多的方法之一,也是目前研究多电子体系结构的常用方法。本文使用Gaussian 09程序优化在分析过程中所用到的分子几何结构进行计算,为了得到在NaOH溶液中木质素二聚体模化物能量最低的构象,采用杂化密度泛函B3LYP/6-31G(d)基组对初始构象进行优化和计算,所有反应物、自由基、产物、过渡态的标准热力学参数是在同一水平上使用频率计算(FRE)命令得到的,并考虑了零点振动能(ZPE)校正,确认过自由基、产物均没有虚频,是势能面上的能量极小点,确认了过渡态有且仅有一个虚频,并对过渡态进行IRC路径计算分析,确认过渡态两端为产物与反应物。同时对木质素二聚体模化物电化学氧化降解反应过程中反应物、产物进行结构优化和频率计算。
对计算结果进行波函数分析,通过提取解析量子化学计算结果,对木质素二聚体进行分子水平的特性分析,绘制静电势图及HOMO和LUMO轨道。其中HOMO轨道是具有电子的最高能级轨道,该轨道的距离原子核最远,受到原子核的引力束缚也最小。因此,该层电子比较容易发生电子跃迁和失去电子。电位越负其还原性越强;而LUMO轨道是离HOMO最近的电子空轨道,LUMO轨道能量越低越有利于得到电子表现出氧化性。
常用于分子结构特征描述的量子化学计算的前线轨道理论:理论认为分子中存在类似于单个原子的“价电子”。这些原子间的价电子就是前线电子,化学反应过程中,原子间的化学键断裂和形成新的化学键起关键作用的电子就是前线电子。最高占据分子轨道(Highest Occupied Molecular Orbital, HOMO)和最低未占分子轨道(Lowest Unoccupied Molecular Orbital, LUMO)统称为前线轨道。
3. 结果与讨论
3.1. 木质素二聚体的电氧化降解
木质素二聚体模化物的裂解主要源于其连接键的断裂,在Gaussian 09的计算中,并不能直接计算出某个特定化学键的键能数据。在计算时将分子从目标键处断裂开,使分子裂解为不同的自由基分子碎片,分别对这些自由基分子碎片进行几何优化,再将这些分子碎片的能量相加。最后将分解后的分子碎片的总能量之和减去分解前分子的总能量,得到的差值即为断裂该化学键所需要的能量,其中键解离能的计算公式为:
(1)
式中,H表示焓,E0表示考虑了零点振动能校正时的键解离能。
在电氧化降解过程中,木质素会形成多种自由基,本文计算了木质素二聚体模化物中六种(D1~D6)连接苯环的化学键以及苯环上R-H (D7~D16)化学键发生断裂的键解离能。在计算解离能前,对木质素二聚体模化物及化学键断裂后生成的自由基进行几何优化,如图3所示,以木质素二聚体模型化合物为例,其会以不同的构象排列出现。通过旋转二面角来构造不同的构象,充分优化并找到能量最小值,选择能量最低及热力学最稳定的分子结构作为本文的研究对象,如图4所示。键解离能是表示化学键强度的一种量度,在电化学氧化降解反应过程中键解离能越小,化学键越容易断裂,反应越容易进行。

Figure 3. Gaussian 09 optimized molecular model of lignin dimer and free radical
图3. Gaussian 09结构优化后的木质素二聚体分子模型及自由基
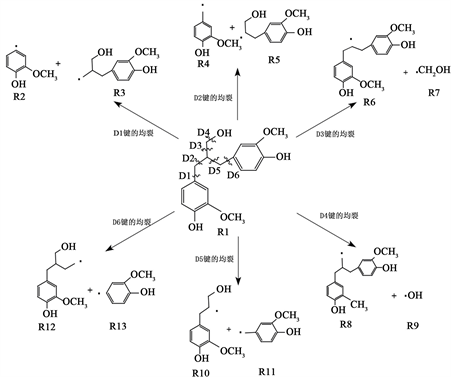
Figure 4. Cleavage path and bond dissociation energy of lignin dimer modulates
图4. 木质素二聚体模化物的裂解路径及键解离能
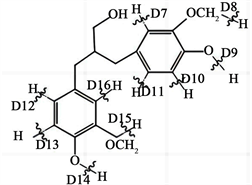
Figure 5. R-H bond cleavage pathway of lignin dimer modulates
图5. 木质素二聚体模化物R-H键断裂途径

Table 1. Bond dissociation energy of R-H bond breaking in dimer modulates of lignin
表1. 木质素二聚体模化物中R-H键发生断裂的键解离能
根据热力学计算数据可知,D1-D6六种化学键键解离能接近,均在380 kJ/mol~430 kJ/mol之间,分别为425.9 kJ/mol、403.1 kJ/mol、405.8 kJ/mol、402.4 kJ/mol、381.2 kJ/mol、421.2 kJ/mol。D7、D10、D11、D12、D13、D16代表的是直接连接在苯环上的氢发生断裂,D8、D15表示的是O-CH3键上的氢断裂,D9和D14则代表的是OH键上的氢断裂,根据表1中计算的数值发现,相较于苯环连接的O-CH3键和OH键上的化学键断裂,苯环上R-H键直接断裂所需要的能量较高。因此,从能垒角度综合计算结果分析,木质素二聚体模化物在电化学氧化降解中优先断裂的是连接两个苯环间的C-C键,断裂生成了木质素单体。随后开始断裂的是与苯环连接的O-CH3和O-H键,而最为稳定、裂解概率最低的是苯环上的C-H键。但由于木质素内部结构及反应过程中影响因素较多,键的断裂顺序和位置还需综合其他因素具体分析(图5)。
3.2. 木质素二聚体反应位点预测
为了探究静电势分布情况,本节计算并绘制了木质素二聚体模化物的静电势图,在静电势图中靠近蓝色部分表示缺电子的区域,靠近红色部分表示富电子的区域(图6)。
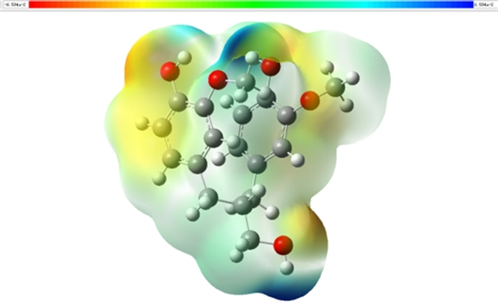
Figure 6. Electrostatic potential of lignin dimer modulates
图6. 木质素二聚体模化物静电势图

Figure 7. (a) HOMO orbitals in lignin dimer modulates; (b) LUMO orbitals in lignin dimer mold molecules
图7. (a) 木质素二聚体模化物分子中HOMO轨道;(b) 木质素二聚体模化物分子中LUMO轨道
20世纪50年代,福井谦一 [19] 总结出前线轨道理论,研究表明,分子附近的电子云根据能量可细分为不同的能带轨道,电子最高占据分子轨道HOMO和电子最低占据分子轨道LUMO影响化合物的很多性质,是决定材料电荷输运性质的最重要的参数之一,也是决定化学反应的关键。为了使电荷传输效率增高,聚合物的HOMO和LUMO应该在合适的位置,以便电子和空穴从金属电极注入到聚合物表面。本研究中使用Gaussian 09对木质素二聚体模化物进行结构优化、频率和单点能计算,然后计算产生电子密度和静电势文件,再由Gaussian View分别绘制木质素二聚体模化物的HOMO和LUMO等值面图。木质素二聚体模化物的HOMO和LUMO电子云分布如图7所示。
HOMO轨道是具有电子的最高能级轨道,该轨道距离原子核最远,受到原子核的引力束缚最小,因此,该层电子比较容易发生电子跃迁和失去电子。电位越负,其还原性越强;而LUMO轨道是离HOMO最近的电子空轨道,LUMO轨道能量越低越有利于得到电子表现出氧化性。计算结果表明木质素二聚体模化物的85轨道为HOMO,轨道能量为−0.19082 eV。木质素二聚体模化物的86轨道为LUMO,轨道能量为0.00543 eV。能量差HOMO-LUMO gap (Egap):0.19625 eV = 18.9353 kJ/mol,从能量计算结果中可以发现木质素二聚体模化物的能量差值较小,分子较为稳定。
HOMO和LUMO轨道图中各原子附近的不规则阴影区域表示电子在HOMO和LUMO轨道出现时其波函数形成的离域空间,离域空间中电子的出现概率较大,电子流动也相对更加剧烈,红色阴影区域表示相位为负的轨道波函数,绿色阴影表示相位为正的轨道波函数。由图7可知,电子在HOMO和LUMO轨道均产生了离域空间,且HOMO轨道图中不规则面积的大小略小于LUMO轨道图中不规则面积的大小,也进一步说明木质素二聚体模化物具有一定的得失电子能力,且得电子能力略大于失电子能力。进一步分析图7中单个原子附近的离域空间,在木质素二聚体模化物的优化构型中,不规则面积主要集中分布在苯环上C原子附近,且苯环连接处的C原子附近的不规则面积占据了HOMO、LUMO轨道图的大部分区域,说明极有可能是木质素二聚体的主要反应活性位点。
3.3 反应路径中反应物、产物的几何构型优化
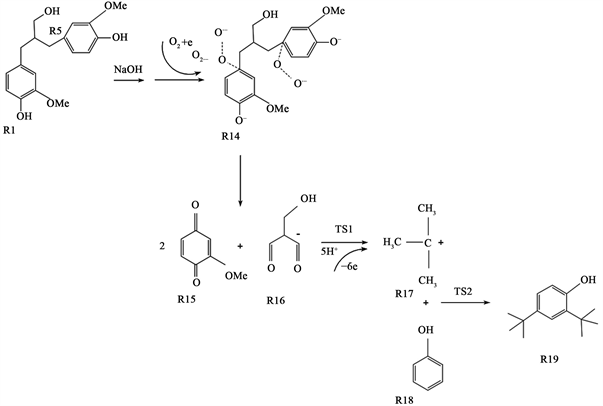
Figure 8. Prediction of reaction path of electrochemical degradation of lignin dimer modulates
图8. 木质素二聚体模化物电氧化降解反应路径推测
根据木质素电氧化降解生成2,4-二叔丁基苯酚的反应机理推测,以木质素二聚体模化物为反应物推测木质素二聚体模化物的反应路径如图8所示,在进行量子化学计算前,对于反应路径中的反应物、中间体及产物进行几何优化。图9列出了木质素电氧化降解路径中的反应物(R)、中间体(IM)和产物(P)优化后的几何构型。
对反应物、中间体、产物进行频率计算,计算结果表明,反应物、中间体、产物都没有虚频,而过渡态均有唯一虚频,验证了各优化结构的合理性。
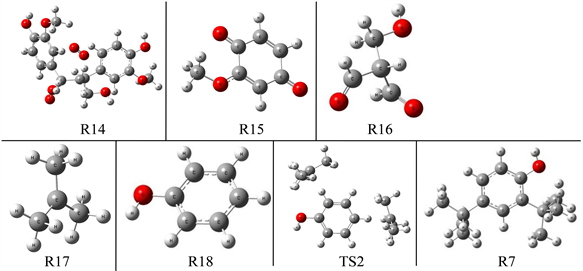
Figure 9. Optimal configuration of R2-R7 in the reaction path
图9. 反应路径中R2-R7的优化构型
图10为木质素二聚体电化学氧化降解反应生成2,4-二叔丁基苯酚的路径反应势能剖面图,由上文原理推测可知,木质素二聚体在NaOH溶液中形成均相碱木质素溶液,木质素二聚体基团中羟基氢质子被夺去,形成木质素负离子基团。水电解生成的氧气在阴极表面得到电子,生成超氧负离子自由基(O2∙−)和氢质子(H+),由于木质素负离子基团和超氧负离子自由基(O2∙−)均带有负电荷,易于被吸附在阳极的表面,发生氧化反应,进而形成木质素过氧负离子自由基;据Magnus Carlsson报道 [20] ,超氧负离子自由基(O2∙−)选择性氧化芳香环取代侧链的C-C键,吸收热量(40.29 kJ/mol)使得侧链断裂,生成的中间产物经过渡态(TS1)被阳极生成的氢质子还原生成异丁烯碳正离子和H2O,同时放出热量。最后,异丁烯碳正离子中间体亲电进攻木质素断裂生成的苯酚结构单元,经过过渡态(TS2)发生亲电加成反应,该反应过程能垒为398.6 kJ/mol,吸收热量0.59 kJ/mol,最终生成产物2,4-二叔丁基苯酚。
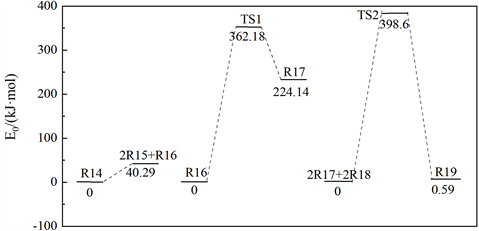
Figure 10. Path reaction potential energy profile of 2, 4-di-tert-butylphenol from lignin dimer
图10. 木质素二聚体生成2,4-二叔丁基苯酚的路径反应势能剖面图
4. 结论
本文从分子层面认识木质素电化学氧化过程以及产物形成的机理,构建木质素理论模型,采用杂化密度泛函B3LYP/6-31G(d)基组对初始构象进行优化和计算,通过键解离能计算、HOMO和LUMO轨道活性点位分析和势能分析等研究木质素二聚体模化物反应路径。研究发现二聚体模化物在电化学氧化降解反应中连接两个苯环的C原子处得失电子能力最强,苯环间的C-C键最易断裂。其次则是与苯环连接的O-CH3和O-H键,而最为稳定、裂解概率最低的则是苯环上的C-H键。其中,苯环间的C-C键断裂形成了木质素单体和异丁烯碳正离子,异丁烯碳正离子与苯酚反应生成产物2,4-二叔丁基苯酚,该反应过程能垒为398.6 kJ/mol,吸收热量为0.59 kJ/mol。本文对木质素电化学氧化降解反应机理的揭示,为木质素电化学氧化耦合电解水制氢的优化提供了理论支撑。
基金项目
上海市教育发展基金会和上海市教育委员会“晨光计划”(18CG51)。