1. 引言
一般来说,不同组织部位的癌症往往是由不同的基因突变引起的,但即使是发生在同一组织部位的癌症,引发癌症的基因突变也可以是不同的。因此,除了传统的临床所见特征如TNM分期 [1]、年龄、家族史等,如何通过组学特征对癌症患者的预后情况进行预测也成为了癌症预后分析的热点研究 [2] [3]。随着基因组学、转录组学技术快速发展,积累了越来越多的基因组学数据,如TCGA、ICGC等大型基因组学数据库,提供了匹配正常细胞基因组的数千个肿瘤的基因组的体细胞突变、体细胞拷贝数变异、DNA甲基化、mRNA表达及患者的临床资料等海量数据。
加权基因共表达网络分析(WGCNA, Weighted Correlation Network Analysis)是用来描述不同样品之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
我们应用加权共表达网络在肺癌中构建差异表达基因的共表达模块,分析模型特征内基因表达与临床特征的相关性,识别枢纽基因(hub gene)。主要的分析步骤包括以下四方面:建立基因加权相关网络;鉴定共表达基因模块;进行模块与临床特征的相关性分析;挑选模块的关键基因。
现已有的研究大多应用单一组学数据进行分析,本文创新点主要是基于多组学多模态的数据,有助于挖掘各模态数据的特征信息,填补单一组学数据中的信息缺失,综合分析患者的相关指标与预后因子之间的关系。同时在WGCNA的基础上运用lasso-logistic等机器学习方法,大大提高了筛选目标特征相关基因的精准率。
2. 相关研究综述
基因组学与影像组学均常用于肿瘤的精准诊疗 [4] [5] [6]。基因组学以分子生物学和信息网络技术从整体上探索全基因组在生命活动中的作用及规律。基因组学可辅助研究肿瘤发生发展的分子机制、发现致癌基因的突变位点和表达通路、分子标记物,但费用和技术要求较高,且获取分子标记物时只能单点单次采集。而肿瘤细胞不仅外部形态不同,内部组织特性也不同,加之所处环境的多样性,肿瘤诊断必须考虑肿瘤差异性和异质性信息 [7],仅凭基因组学难以全面描述肿瘤组织的时空异质性。影像组学则是从影像中提取定量特征,用于肿瘤的早期诊断、分期和预后预测等 [8] [9] [10]。影像组学在诊疗过程中能随时跟踪肿瘤的发展状况,但特异性较差,获取重要影像标志物时,生物和分子机制的解释性不足。肿瘤识别是典型的小样本问题,基于影像提取大量底层特征进行肿瘤分类识别时,计算时间长,分类识别精度也有限。另外,临床中要求所提取的特征是稳定、可重复的,但不同的采集设备、参数设置以及特征选择方法均会使所获取的影像特征存在差异,故基于大量底层特征获取影像标记物时,需克服图像采集和特征计算标准化的问题。
国内外在癌症预后领域已有不少研究成果,杨娟等 [11] 采集128例肝内胆管癌患者临床资料,使用Logistic回归分析法筛选预后危险因素以便预测患者术后复发风险,Zhu等 [12] 设计了监督式主成分回归法,将基因表达数据与病理图像数据进行融合,用于肺癌的生存期预测。这种前端融合一定程度提升了生存预测性能,但仅融合了数据低维的原始特征,包含大量噪声和冗余信息。Mobadersany等 [13] 综合考虑组学信息和病理图像信息,对于癌症生存期的预测提出了生存卷积神经网络(SCNN)法,用于多形性胶质细胞瘤的生存期预测。Lai等 [14] 开发了一种结合基因表达异质性数据和临床数据的双峰深度神经网络预测肺癌患者的5年总体生存状态(AUC = 0.816)。
随着各种癌症组学数据的积累,诸多研究工作开始尝试从多模态、多任务的角度出发,融合多组学和病理图像数据,进一步改进癌症生存期预测。Zhang 等 [15] 提出了一个基于多核学习框架的多模态数据前端融合模型,成功融合基因表达、拷贝数变异、甲基化等多种组学数据,进行了癌症预后的预测。多模态数据融合算法充分考虑并有效提取了模态间的关联性,因此这类基于多模态数据融合的模型在癌症生存期预测中均取得了不错的效果。
3. 基于加权共表达网络肺癌预后模块鉴定
3.1. 肺癌预后模型结构说明
我们将建立肺癌预后模型工作分为三个模块:数据模块、特征基因选择模块和预后模型建立模块。第一个模块为数据模块,主要完成数据获取与预处理的功能。第二个模块为特征基因选择模块,在该模块中,我们要在全基因组数万个基因中剔除与肺癌患者预后相关性不大的基因,筛选出与肺癌患者预后密切相关的少数基因。第三个模块为预后模型建立模块,在该模块中,我们的主要任务是用四种机器学习算法来构建预测肺癌患者总生存时间是否超过5年的预后模型,并对四种模型的分类的AUC值进行计算和比对,挑选效果最好的预后模型。图1展示了构造模型的结构层次图。
3.2. 实验数据获取与预处理
TCGA (The Cancer Genome Atlas,癌症基因图谱)是由美国国家癌症研究所和美国国家人类基因组研究所于2006年联合启动的项目。该项目收录了人类癌症的各种数据,如临床数据、miRNA表达数据、甲基化等数据。
本文所采用的数据来自于TCGA数据库,主要使用肺癌患者的基因表达量、拷贝数变异、外显子基因表达量和临床数据,其中临床数据主要使用生存信息。从UCSC官网(https://xenabrowser.net/heatmap/)中下载TCGA肺癌基因表达量、拷贝数变异、外显子基因表达量和临床数据。在对数据进行分析之前,我们首先剔除掉缺失关键信息的样本,并且只采用同时含有三个组学信息的癌症患者样本,最终得到995个样本的基因数据,清洗数据以后对数据进行标准化处理,同时根据临床数据中的总生存时间(OS.time)将患者按生存时间是否超过5年分为OSfive = 1和OSfive = 0两类,其中OSfive表示患者五年生存期,OSfive = 1即为患者生存时间超过5年。
3.3. 加权共表达网络分析
3.3.1. 加权共表达网络概述
加权共表达网络(Weighted Gene Correlation Network Analysis, WGCNA)可以将基因网络根据表达相似性划分成不同的模块,并分析模块与特定表型之间的相关关系。在WGCNA算法 [16] 中,我们用节点代表基因,用节点与节点之间的连线代表基因表达相关性,邻接度用来表示节点之间的关系强弱,网络内的所有基因的邻接度可以形成一个邻接矩阵。在无尺度网络中,只有极少数节点与很多节点有关,因此,一个模块中有许多基因但关键基因只有几个,我们选择最佳加权系数使基因服从无尺度网络分布。
3.3.2. 加权共表达网络构建
传统方法中描述两个基因之间的关联程度一般会指定一个筛选阈值,但这种方法会丢失基因的变化趋势信息,为了解决这些问题,加权共表达网络对基因表达值之间的相关系数取β次幂,对于基因i和j,相关系数为
,取β次幂后得到
,可以得到
,最终将基因间相关性的强弱的差别放大,这样的好处是使强弱关系更为分明有利于后续聚类识别。
加权共表达网络的构建基于RStudio软件中的“WGCNA”函数包 [17] 来实现,加载“WGCNA”包,使用“goodSamplesGenes()”函数检查数据是否有异常基因,使用“hclust()”函数绘制聚类图,再应用“cutree()”函数剔除离群样本,获得较为一致的基因表达数据。然后使用“pickSoftThreshold()”函数进行自动网络拓扑分析,依据无尺度网络原则确定软阈值参数(β),根据图2选择合适的β保证拟合优度在0.9以上,将相关性矩阵转化为邻接矩阵,构建一个趋向无尺度网络的加权共表达网络。
3.3.3. 肺癌预后模块的选择
将选择的β值代入“blockwiseModules()”函数,设置最小模块基因数、模块合并阈值等参数,划分模块并合并相似模块。然后计算模块特征向量和临床性状之间相关系数矩阵,并对相关系数进行检验。图3将相关系数矩阵进行热力图可视化,在图4中挑选p值小于0.05且相关性较高的模块作为备选模块。
3.3.4. 肺癌预后特征基因的选择
基因的模块身份(Module Membership, MM)用于描述基因在所有样本中的表达谱与某个特征向量基因表达谱的相关性,即对module eigengene进行相关性分析就可以得到MM值,所以MM值本质上是一个相关系数,如果基因和某个module的MM值为0,说明二者根本不相关,该基因不属于这个module,如果MM的绝对值接近1,说明基因与该module相关性很高。

Figure 2. Determining soft threshold parameters based on scale-free network principle
图2. 依据无尺度网络原则确定软阈值参数

Figure 3. Module division and module merging diagram
图3. 模块划分与模块合并图
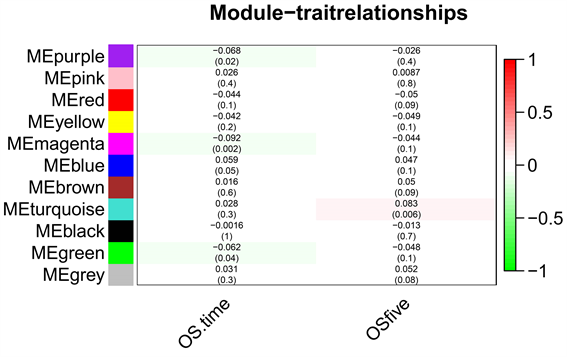
Figure 4. Module correlation coefficient matrix heatmap
图4. 模块相关系数矩阵热力图
基因显著性(Gene Significance, GS)用于描述模块内基因与OSfive关联程度,将基因的表达量与对应的表型数值进行相关性分析,最终的相关系数的值就是GS。GS反映出基因表达量与表型数据的相关性,GS越高说明基因在预后方面越有意义。
根据图5结果,我们挑选备选模块中GS绝对值大于0.1且MM绝对值大于0.8的基因,取备选模块中筛选出的基因的并集作为所选择的特征基因。

Figure 5. Module and gene expression correlation plot
图5. 模块与基因表达相关性图
3.4. 结果
经过筛选后我们得到51个特征基因,考虑到所选基因之间可能存在多重共线性问题,我们用方差膨胀因子(Variance Inflation Factor, VIF)方法处理存在严重共线性的特征基因,应用R软件中的car包,使用“vif()”函数将VIF数值大于100的特征基因剔除,最终获得39个特征基因。在这一环节我们通过WGCNA算法完成了特征基因选择。
3.5. 预测模型构建
3.5.1. 逻辑回归模型
逻辑回归(Logistic Regression,LR)算法是在线性回归模型的基础上,添加sigmoid函数来完成映射,将连续值转化为(0, 1)之间的一个概率值,通过这个概率值我们可以解决分类问题。本文所解决的肺癌分类问题为二项分类问题,因此选择二项逻辑回归模型作为分类模型,二项逻辑回归模型的条件概率分布表示如下所示。
(1)
(2)
其中,
是输入变量,
是输出变量,w叫做权重,b叫做偏置量。
3.5.2. 支持向量机算法
支持向量机(Support Vector Machines, SVM)是一种强大的分类器构建方法,它的学习策略为间隔最大化,即找寻一个可以最大化将训练数据分隔开的超平面。在样本空间中,划分超平面可以通过如下方程来描述:
(3)
其中w为法向量,决定了超平面的方向;b为位移项,决定了超平面与原点之间的距离。对于所有训练数据,w和b应该满足以下两个不等式:
(4)
(5)
这些满足
条件的
则称为支持向量。所以支持向量机可理解为一个求解二次凸优化问题,如下所示:
(6)
(7)
3.5.3. K近邻
K近邻法(K-Nearest Neighbor, KNN)是一种常用于分类的算法,K近邻利用距离来区分类型的思路是:通过离测试点最近的k个已知点的类型来决定测试点的类型,其中k是人为设定的数值,k的取值对模型的好坏有非常重要的影响。
在训练集中,所有的数据和数据所对应的分类标签已知。每当有一个新的测试数据输入时,KNN算法会计算测试数据与每一个训练数据之间的距离,并将距离进行排序。然后KNN算法会找出前k个与新数据点最近的训练集样本点和它们相应的标签,该测试数据对应的类别就是这k个数据中出现次数最多的分类。
KNN是一种懒惰的学习算法,训练数据不需要执行任何泛化,同时,两个数据点的距离计算至关重要,不同的距离计算公式对KNN的模型分类效果产生巨大影响。在实际中两种最常用的距离计算公式是欧式距离和曼哈顿距离,计算公式如下:
欧式距离:
(8)
曼哈顿距离:
(9)
3.5.4. 随机森林
随机森林(Random Forest)是一种比较新的机器学习模型集成学习方法,随机森林对多元共线性不敏感,结果对缺失数据和非平衡数据比较稳健。随机森林是用随机的方式建立一个森林,森林由很多的决策树组成,并且每一棵决策树之间是没有关联的,得到随机森林模型后,当新样本进入时,随机森林的每一棵决策树会分别进行判断,对于分类问题通常使用投票法,得到最多票数类为最终模型输出。
若训练集大小为N,对于随机森林中每棵树而言,随机且有放回地从训练集中抽取N个训练样本作为该树的训练集。随机森林算法通过随机抽取训练样本作为每棵树的训练集,可以使得构成森林的每棵树的训练集都不一样,进而减少不同树之间的相关性,使分类效果更好。
通过设计多个决策树并将它们的预测结果相结合起来,随机森林大大降低了过拟合的风险,并且使得构建出的随机森林具有很好的抗噪能力。
4. 结果
4.1. 肺癌预后模型的实现
在上一步的工作中,我们利用WGCNA算法从全基因组上万个基因中筛选出了与肺癌患者预后生存密切相关的39个特征基因。图6的结果是运用主成分分析(PCA)做出的主成分碎石图,选取主成分所占百分比前六的六个主成分,通过主成分分析剔除测试集中的异常样本,见图7。我们使用机器学习算法构建预测肺癌患者总生存时间能否超过5年的预后模型,通过这个二分类模型可以更好的辅助医生将不同患者分入不同的危险组别之中。

Figure 7. Variables on dimensions 1 and 2
图7. 变量在主成分1和2上表示
在本节工作中,我们同时使用逻辑回归、支持向量机、K近邻、随机森林四种机器学习算法构造出四个不同的肺癌患者预后模型,通过对四种预后模型效果进行比对,筛选出一个最好的预后模型。由于肺癌患者基因数据有限,为得到可靠模型,在训练模型时使用10折交叉验证,通过交叉验证方法在一定程度上避免过拟合,使预后模型具有更好的泛化能力。
4.2. 预后模型分类效果
在实验过程中,我们对所有模型都采用10折交叉验证,通过将数据集按照8:2划分训练集和测试集,通过多次划分多次训练,实验结果取十次实验的平均值来提高模型的泛化能力,在这一环节我们使用AUC指标来评价模型的好坏。
ROC曲线如图8所示,其中红色曲线为逻辑回归ROC曲线,蓝色曲线为支持向量机ROC曲线,紫色曲线为K近邻ROC曲线,绿色曲线为随机森林ROC曲线。
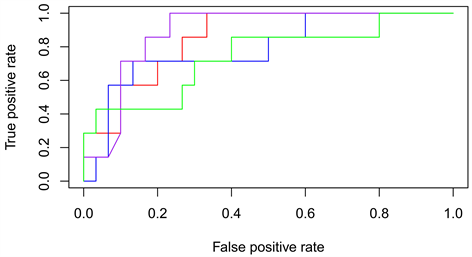
Figure 8. ROC curves of four prognostic models
图8. 四种预后模型的ROC曲线
表1展示了四种机器学习算法在预测肺癌患者5年总生存时间的AUC值,从表1中可以看出,四种模型的AUC值都在0.7以上,其中KNN算法构建的模型效果最佳,AUC值达到0.888。

Table 1. Classification results of prognostic model based on characteristic gene construction
表1. 基于特征基因构建的预后模型分类结果表
5. 结论
本文考虑肺癌患者基因的多组学多模态数据,通过TCGA数据库获取相关组学数据,对数据进行预处理后,基于加权共表达网络算法筛选出来39个特征基因,运用机器学习算法构建了肺癌患者预后预测模型,对所有的预后模型效果进行比较,得到使用基于KNN算法构建的预后模型效果最好,AUC值达到0.888,能够较为准确地判断肺癌患者生存时间是否超过5年。