1. 引言
随着工业化进程的不断加深,我国在经济发展上取得卓越成果的同时,也面临着诸多日益严重的环境问题,重金属污染即是其中之一。排放到环境中的重金属难以被降解,通过空气、水、土壤等途径进入食物链,最后通过富集作用不断累积后进入人体 [1] 。常见的重金属污染物主要包括铅(Pb)、砷(As)、汞(Hg)、锌(Zn)、镉(Cd)等。这些重金属污染物均能对人体造成不同程度的损害。其中,铅对人体的各组织器官均有毒性。对于成人,常导致心脏衰竭、腹痛、贫血、高血压、关节疼等症状;对于幼儿则会产生神经和脑功能等不可逆的损伤,严重者致痴呆。而且,即使经过治疗使体内的血铅含量回复正常水平,造成的组织器官损伤已无可挽回 [2] 。
吸附法是治理重金属污染的常用方法 [3] ,而选择合适的吸附剂是吸附法的关键,在吸附效果、经济性、环保性等方面均需要斟酌。天然粘土矿物产量大,经济环保,其吸附能力也很有潜力,国内外诸多学者已对此进行了实验和研究 [4] [5] 。
高岭土广泛存在于土壤和沉积物中,是一种常见的天然粘土矿物,其有效成分为高岭石,作为重金属污染物的吸附剂有很大的潜力 [6] [7] 。针对高岭石对重金属吸附的研究,在宏观实验方面,已有大量的研究成果 [6] [7] [8] [9] [10] ;而在微观方面的研究较少 [11] [12] ,本文采用基于量子力学的第一性原理,从微观角度对高岭石(001)面吸附铅原子的吸附特性进行了计算和讨论,为高岭石吸附重金属原子的应用提供理论依据和支持。
2. 计算方法
本文使用平面波和投影缀加波的VASP软件(Vienna ab-inito simulation package)进行第一性原理计算 [13] 。高岭石模型使用2 × 2的周期性超原胞结构,原子个数为68个,对所有原子进行弛豫时采用PBE型交换关联势 [14] 。在计算过程中,全部的原子位置都按照Hartree-Fock [15] 力进行弛豫,布里渊区积分使用Monkhorst-Pack [16] 方法。平面波函数的截断能设置为450 eV,k点网格设置为3 × 3 × 1可以保证计算的精确度 [17] 。在弛豫计算过程中,核运动收敛精度设置为小于0.01 [17] 。高岭石的晶体结构是三斜晶系,优化后的晶格常数为a = 5.160 Å,b = 5.160 Å,c = 7.602 Å,α = 81˚,β = 89˚ and γ = 60.18˚与实验结果符合很好 [18] ,计算模型可靠。计算模型分别考虑了高岭石(001)和(00-1)面各有三种对称吸附位置 [18] ,即顶位(top)、桥位(bridge)和洞位(hollow),对于(001)面分别指在氧原子的正上方的位置,两个氧原子中间的上方位置,三个氧原子正中间位置,如图1所示;对于(00-1)面分别指在硅原子的正上方的位置,两个硅原子中间的上方位置,六个硅原子正中间位置,如图2所示。真空层厚度取20 Å。
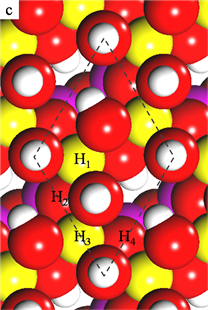
Figure 1. Top view of the kaolinite (001) surface with (a) top adsorption sites, (b) bridge adsorption sites, (c) hollow adsorption sites. The colors of H, Al, O and Si atoms are white, yellow, red and pink, respectively
图1. 高岭石(001)面三种高对称吸附位置:(a) 顶位,(b) 桥位,(c) 洞位;图中白色,黄色,红色,粉色分别表示H,Al,O,Si原子

Figure 2. Top view of the kaolinite (00-1) surface with (a) top adsorption sites, (b) bridge adsorption sites, (c) hollow adsorption sites. The colors of H, Al, O and Si atoms are white, yellow, red and pink, respectively
图2. 高岭石(00-1)面三种高对称吸附位置:(a) 顶位,(b) 桥位,(c) 洞位;图中白色,黄色,红色,粉色分别表示H,Al,O,Si原子
针对上述各高对称吸附位置,分别考虑其对铅原子的吸附性质,建立高岭石吸附铅原子模型后进行计算。高岭石和铅原子之间的吸附能用以下公式计算 [19] :
(1)
上式中,
、
、
分别表示单个铅原子、洁净高岭石表面、吸附体系的平衡能量。即体系的吸附能等于铅原子和高岭石表面的能量之和减去吸附体系的能量。
为了更进一步的了解铅原子在清洁高岭石表面吸附的本质特点,本文还计算了铅原子和高岭石(001)面原子的差分电荷密度和态密度,体系差分电荷密度按以下公式计算 [19] :
(2)
上式中,
、
、
分别表示吸附体系、单个铅原子、洁净高岭石(001)面的电子密度。即体系差分电荷密度等于吸附体系的总电荷密度减去单个铅原子和高岭石表面的电荷密度,在三维差分电荷密度图中,蓝色区域表示电荷积聚,黄色区域表示电荷减少。
3. 结果分析
3.1. 高岭石吸附单个铅原子
本文分别对于高岭石(00-1)面和(001)面,均使用三种高岭石吸附铅原子的模型进行模拟计算。经过计算,在高岭石(001)面,弛豫后的铅原子稳定吸附在高岭石(001)面的顶位和桥位两个高对称吸附位,如图3所示。根据吸附能计算公式(1),铅原子在高岭石(001)面的顶位时,具有吸附能约为0.96 eV,在桥位时吸附能约为1.07 eV;然而对于高岭石(00-1)面,单个铅原子的最大吸附能仅为0.10 eV,与(001)面相比吸附作用较小,故本文对其吸附性不再做讨论。
为了更深入了解高岭石(001)面与铅原子的相互吸附作用,本文对铅原子在顶位和桥位的三维差分电荷密度和态密度分别进行了计算,如图4所示。为了对比分析吸附前后的变化,本文还计算了自由铅原子、洁净高岭石表面、以及吸附体系的态密度。
对比分析顶位吸附前后铅原子s、p轨道态密度(图4(a))可以看出,吸附后的铅原子s轨道和p轨道相对于费米面分别下移了约0.3和0.2 eV。同时铅原子s和p轨道在吸附后峰值有所降低,都说明了铅原
Figure 3. Pb atoms adsorbed on top (a) and bridge (b) site
图3. 铅原子在顶位(a)和桥位(b)吸附后的形态

Figure 4. Contrast diagram of the difference electronic density and state density of Pb and O atoms of kaolinite (001) surface before and after adsorption
图4. 铅原子和高岭石(001)面顶位氧原子在吸附前后的差分电荷和态密度对比图
子的s和p轨道的电荷转移并重新分布。图4(b)展示了铅原子在顶位吸附后,与其相互作用的高岭石表面氢和氧原子的态密度变化。从图中可看出高岭石表面氧氢原子的s和p轨道能量下移,并与氢原子在能量区间−10~−7.5 eV重合。从图4(a)的差分电荷密度插图中可以清楚的看出,吸附的铅原子周围电荷减少,而高岭石表面与铅原子相互作用的氧原子周围有电荷聚积区域,差分电荷密度图也说明了相互作用后电荷的转移和重新分布,吸附属于稳定的化学吸附。
同样我们也计算了铅原子在桥位的差分电荷密度和态密度(图4(c)和图4(d)),从图中看出,吸附后的铅原子s轨道和p轨道相对于费米面分别下移了约0.6和0.3 eV。同时在吸附后,轨道峰值降低较大且展宽明显,与顶位吸附比较更为明显。其次高岭石表面氢氧原子s和p轨道能量下移更多,同时在−7.5 eV出现一个新的峰值,并与铅原子s轨道相重合。最后从图4(c)差分电荷密度可看出,铅原子表面与铅原子相互作用的两个氧原子周围有电荷聚积区域,说明了相互作用后电荷的转移和重新分布,吸附属于稳定的化学吸附。
3.2. 高岭石吸附多个铅原子
在计算高岭石吸附单个铅原子的基础上,本文还讨论了高岭石(001)面两种高对称吸附位置吸附多个铅原子问题。计算所选取的高岭石是2 × 2的超原胞,对于顶位分别计算了铅原子的覆盖度为Θ = 0.08、0.25、0.33、0.50、0.75、1.0 ML (monolayers)这六种情况;对于桥位分别计算了铅原子的覆盖度为Θ = 0.125、0.25、0.50、0.75、1.0 ML这五种情况。
对上述不同覆盖度的铅原子分别进行计算,高岭石和铅原子之间的吸附能用以下公式计算 [19] :
(3)
上式中,
、
、
分别表示单个铅原子、洁净高岭石表面、吸附体系的平衡能量。
指的是铅原子的个数。也就是说体系的吸附能等于铅原子和高岭石表面的能量之和减去吸附体系的能量,除以吸附铅原子的个数。根据公式(3),计算得到了高岭石表面两种高对称吸附位置顶位和桥位对不同覆盖度铅原子的吸附能,结果如表1所示。
对于铅原子在顶位吸附,当Θ = 0.08 ML时,铅原子竖直稳定吸附在高岭石表面(001)的顶位,能量为0.96 eV。当Θ = 0.25,0.33,0.50,0.75和1.0 ML时,多个铅原子也可吸附在高岭石表面,吸附能分别为2.29,2.53,2.76,30.3和3.37 eV,即在0.08﹤Θ ≤ 1.0 ML范围内,吸附能随着覆盖度的增大而增大。另一方面,随着吸附的铅原子个数的增加,越来越多的铅原子出现了不同程度上偏离顶位的现象。但当Θ = 1.0 ML时,之前的偏移现象几乎消失,各个铅原子于顶位的偏移很小。此时得到最大吸附能3.37 eV。分析其原因,铅原子之间的相互作用影响了其在高岭石表面的吸附位置和吸附能,随着吸附的铅原子的个数的增加,铅原子间相互作用也更加复杂,所以在出现了铅原子不同程度偏离顶位的现象。而当Θ = 1.0 ML时,铅原子间的相互作用取得了平衡,因此偏离顶位的现象得到了改善。
对于铅原子在桥位吸附时,Θ = 0.125,0.25,0.33,0.50,0.75和1.0 ML时,吸附能分别为1.07,2.05,2.71,2.77和2.92 eV,可以看出随着Θ的增加,吸附能也逐渐增大。同时,随着覆盖度的增加,铅原子更多的吸附在顶位而不是桥位,当Θ = 1.0 ML时,全部铅原子均吸附在顶位。因此高岭石吸附多个铅原子时,顶位的稳定性更好。
为了更好的说明铅原子和高岭石(001)面的相互作用,本文还分别计算了不同覆盖度铅原子在高岭石(001)表面顶位,桥位吸附后的垂直距离和层间距变化,其中hPb-O是指铅原子距离高岭石(001)面O原子层的垂直距离,dPb-O是指铅原子距离高岭石表面的O原子的最短键长,Δd12是指高岭石第一层(H原子层)和第二层(O原子层)之间距离的相对变化量,根据公式
得到,d12和d0分别表示吸附后和吸附前高岭石第一层和第二层的层间距,结果为正值说明吸附后层间距增大,反而说明减小。同样Δd23是指高岭石第二层(O原子层)和第三层(Al原子层)之间距离的相对变化量。从表2可以看出,铅吸附在顶位和桥位后,随着覆盖度的增大,其与氧原子层表面的垂直距离均呈递增的趋势。从铅原子与高岭石表面的垂直距离的逐渐增加但高岭石表面对其的吸附能也在逐渐增加的现象也可以证实铅原子之间的相互作用对吸附能产生了影响。
从表2中Δd12和Δd23数据可以看出,铅原子在高岭石表面(001)面顶位吸附后,第一、二层和第二、三层的层间距在减小,说明随着吸附个数增多,高岭石分子结构压缩趋势在变缓。Δd12和Δd23分别从3.19%,4.15%逐渐变化至1.52%,2.82%。与之相反的是,铅原子随着在桥位的吸附个数的增多,第一、二层的层间距在增大,从膨胀到压缩状态变化,而第二、三层的层间距呈减小趋势。同样说明吸附个数对高岭石结构的影响较大。最后我们还计算了铅原子吸附后与其高岭石氧原子的成键距离,从表中可以看出,顶位和桥位随着吸附铅原子个数的增多都呈增长趋势,这与铅原子之间相互作用有关。

Table 1. Adsorption energy of different coverage Pb atoms on two adsorption sites of kaolinite (001) surface
表1. 不同覆盖度铅原子在两种不同高对称吸附位置的吸附能
Table 2. The calculated changes of the adsorbate height (hPb-O), the interlayer relaxation (Δd), and the bond length (dPb-O) for different coverage of atomic Pb adsorption on kaolinite (001) surface
表2. 高岭石吸附不同覆盖度铅原子的垂直距离(hPb-O),层间距(Δd)和最短键长(dPb-O)变化
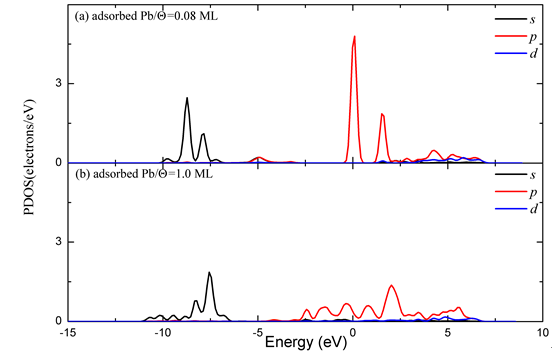
Figure 5. Contrast diagram of the state density of several Pb atoms and kaolinite (001) surface before and after the adsorption of the highest level atom
图5. 多个铅原子及高岭石(001)表面最高层原子在吸附前后的态密度对比图
为了深入了解高岭石表面(001)吸附单个和多个铅原子的不同,本文还计算了顶位吸附Θ = 0.08 ML和Θ = 1.0 ML的态密度(图5)。图5(a)和图5(b)分别为单个和多个铅原子在吸附后的s、p、d轨道态密度。对比分析不同覆盖度下铅原子s和p轨道态密度可以看出,高覆盖度的铅原子s轨道和p轨道比单个铅原子的峰值有明显降低和幅度展宽,这些特征说明了多个铅原子吸附时,其s和p轨道有更多的电荷转移,相互吸附作用更强。
4. 结论
本文采用基于密度泛函理论的第一性原理计算研究了高岭石(001)和(00-1)面与铅原子的相互作用,得到了铅原子在高岭石(001)面高对称位置的吸附能、稳定吸附结构及其吸附后的层间距、态密度和差分电荷密度的变化等性质。计算结果表明铅原子与高岭石(001)面之间形成作用较强的化学吸附,两种高对称吸附位置顶位和桥位对单个铅子的吸附能分别0.96和1.07eV,其中桥位吸附能量最大,吸附最稳定。其次,本文还计算了不同覆盖度铅原子在高岭石(001)面的吸附行为。随着Θ的增大,高岭石(001)面两种吸附位置对铅原子的吸附能均成逐渐增大的趋势,当Θ = 1.0 ML时,二者具有最大吸附能分别为3.37和2.92 eV。最后,对吸附后铅原子和高岭石(001)面的态密度和差分电荷密度进行了对比分析,吸附后电荷的转移和重新分布说明了吸附作用较强,属于化学吸附。同时还计算了铅原子与高岭石表面的垂直距离以及高岭石分子结构层间距的变化,说明较强的相互作用对高岭石分子结构有较大的影响。
基金项目
国家自然科学基金项目(No.41702317)和中国岩石力学与工程学会“青年人才托举工程”项目2015-2017联合资助。