1. 引言
计算机辅助药物设计(Computer-aided drug design, CADD)的发展是随着结构生物学与计算化学的进程的加快,使得CADD技术在整个药物设计与开发中起着越来越重要的作用 [1] [2] [3] 。如今CADD技术介入药物研究最多的领域是针对某一个药物的作用靶标 [4] ,在已经开发的化合物数据库搜寻筛选中,找到对该靶标具有特异作用的化合物。这些数据库包括以往的基于化学合成的商品化合物数据库的搜索以及近年来国内研究机构已经授权代理销售的TCMD (中药化学数据库) [5] 和CNPD (中国天然产物数据库) [6] 等。显然CADD技术一方面大大降低药物开发成本,节省有限的实验资源,另一方面大大缩短药物开发周期,加快新药的发现与制备速度成为国内外目前十分活跃的研究领域。2001年反向分子对接(inverse docking)概念的提出 [7] ,以其特有的高效便利等特点,为药物靶点的发现和确认提供了新的思路。基于药物靶分子虚拟筛选构成的两个方面,分为基于受体结构的筛选和基于配体结构的筛选,如INVDOCK、TarFis Dock、PharmMapper [8] 等软件工具被相继开发被提供后,越来越多的天然活性成分的靶点得以识别和验证,使进一步设计新作用特点的先导化合物或治疗药物高通量筛选成为可能,对新药创制具有源头创新意义。
具有重要生理功能和新颖结构的天然产物的发现和研究,是新药研究的主要来源。现有天然产物研究靶点信息学手段总结如图1所示。
2. 方法和原理
2.1. 天然产物E-低聚白藜芦醇化合物杀菌机制靶点筛选以及分子对接模拟实验
2.1.1. 模型构建
以前期我们从植物化学研究发现的、来自芍药科牡丹籽中分离得到的低聚白藜芦醇类近100多种中

Figure 1. Existing natural products research target informatics means
图1. 现有天然产物研究靶点信息学手段
[9] [10] [11] [12] ,唯有E-低聚白藜芦醇天然化合物1和化合物2,具有极强的抗革兰氏阳性菌和抗耐甲氧西林金黄色葡萄球菌(Methicillin-resistant Staphylococcus aureus, MRSA,分离株)的灭杀作用,详见化合物结构式如图2所示。实验采用药敏法体外筛选,结果显示化合物1的MIC值为8 µg/mL,其MBC值为16 µg/mL;化合物2的MIC值为16 µg/mL,其MBC值为64 µg/mL当用万古霉素做阳性对照时,万古霉素48 h后产生了耐药性,因此断定化合物1和2具有抗生素样的新的抗生素先导化合物,具有替代抗耐药性抗生素潜能,这一研究具有重大科学理论意义。为取得该理论研究的突破,进一步对E-低聚白藜芦醇化合物杀菌机制靶点进行筛选以及分子对接的模拟实验。实验的模型构建,即以化合物1和2以及阳性对照的万古霉素共三个化合物的分子结构提交给PharmMapper server,经过对他们结构的优化,生成多种构象,选择全体蛋白靶标库,其他参数选为默认。PharmMapper sever对配体小分子与其数据库中药效团自动匹配, 然后按匹配的打分高低排序。
2.1.2. 小分子探针的预处理
化合物1和2以及阳性对照的万古霉素共三个化合物的分子结构式如下图2所示。
然后利用DS3.0里的Prepare Ligand模块对这三个化合物进行预处理,结果显示化合物1共有十二种可能的空间构象,化合物2共有三十六种可能的空间构象,万古霉素则仅有两种可能的空间构象。然后得到3个不同化合物最优构象如下图所示。



2.1.3. 受体PBP2a靶蛋白的预处理
本次实验拟选择来自MRSA的PBP2a蛋白作为对接受体蛋白。在PDB蛋白数据库中找到ID为4CJN的靶蛋白下载并在DS3.0中打开,如图3所示。
通过对PBP2a靶蛋白的预处理并定义其受体蛋白的活性位点于10号活性区域,详见如图4所示。
2.1.4. 利用Discovery Studio 3.0对筛选结果进行分子对接验证筛选结果的实验操作
首先将受体蛋白与万古霉素进行分子对接,利用DS的LibDock模块进行模拟分子对接,模拟分析后可以得到分子对接绝对自由能(Absolute Energy)、对接姿式数(Conf Number)、相对自由能(Relative Energy),以及LibDock综合打分(LibDock Score)等参数值,结果详见如表1所示。其中,该抑制剂与位点对接所需的能量是以相对自由能和绝对自由能来表征的,该对接位点的对接越紧密则绝对自由能和相对自由能越低,即抑制剂对该靶蛋白的抑制作用越强。抑制剂与该位点作用的可能性越大则对接姿势数匹配度越高。LibDock模块综合绝对自由能、相对自由能和对接姿式数对该对接姿式的打分并给出LibDock综合打分;LibDock综合得分越高,表明抑制剂对该位点的抑制作用越强。分子对接二维选取对接匹配度最高、对接姿势最好的进行评价。结果如图5所示。
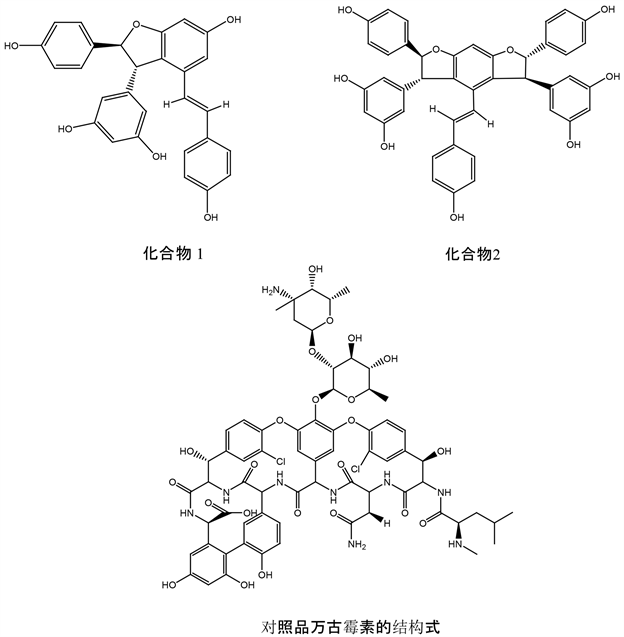
Figure 2. The structures of compounds 1 and 2 and vancomycin
图2. 化合物1和2及万古霉素的结构式

Figure 3. The 2D structures of PBP2a target protein
图3. PBP2a靶蛋白二级结构图示
Figure 4. The defined active site of the PBP2a receptor protein
图4. 定义的PBP2a受体蛋白的活性位点
 (a) The docking result of vancomycin with PBP2a (a) 万古霉素与PBP2a的对接结果
(a) The docking result of vancomycin with PBP2a (a) 万古霉素与PBP2a的对接结果  (b) The docking result of Compound 1 with PBP2a (b) 化合物1与PBP2a的对接结果
(b) The docking result of Compound 1 with PBP2a (b) 化合物1与PBP2a的对接结果  (c) The docking result of Compound 2 with PBP2a(c) 化合物2与PBP2a的对接结果
(c) The docking result of Compound 2 with PBP2a(c) 化合物2与PBP2a的对接结果
Figure 5. 2D diagram of three compound molecular dockings for receptor PBP2a protein. (a) vancomycin, (b) Compounds 1, (c) Compound 2
图5. 3个化合物与受体蛋白PBP2a的2D分子对接图。(a) 万古霉素,(b) 化合物1,(c) 化合物2

Table 1. Docking parameter table of ligand and PBP2a
表1. 配体与PBP2a的对接参数表
3. 结果与讨论
3.1. 生成配体-蛋白复合物对接模型构建与模拟结果分析
生成的配体-蛋白复合物对接模型评价方法PharmMapper服务器打分中fit score可以反映配体构型与药效团模型的叠合情况,因此本实验从上述表1的配体与PBP2a的对接参数表明,化合物1和2与阳性对照万古霉素对PBP2a的抑制作用相比,前者抑制活性稍弱于抗生素的万古霉素,这个结果与体外抑菌效果实验数据是吻合一致的。对比表1各项参数进一步分析可发现,如果从分子对接后形成的内能和对接姿态及位点综合反应抑菌效果的话,化合物1抗菌潜能应最佳;我们的杀菌试验研究也相应表明,两个E-低聚白藜芦醇化合物杀菌显著,但万古霉素48 h已产生耐药现象。这个分子对接揭示了基本结构与分子整体性及药效团与化学结构的药效规律。他们可总结为万古霉素分子基本结构与整体刚性强,分子对接构像难以发生变化,绝对自由能就高,在这种情况下,药物由优势构象转变为药效构象时分子内能增高,如表1中的万古霉素的高的Absolute Energy;另一方面万古霉素化学结构药效团多,作用结合位点最多,在对接姿式上又占有绝对优势,如表1中的万古霉素的高的Conf Number;所以万古霉素得分最高,活性最好;而小分子探针E-低聚白藜芦醇化合物分子的空间位置和姿态的改变容易,参与对接的形成有利 [13] ,对接时可提供药物维持的最优构象和药效构象,即药物与受体间的作用力提高,如表1中的化合物1的LibDock Score (得分132.226),抗菌效果因而较强。然而如表1中的化合物2分子结构整体药效团少,对接位置和姿态受到限制,因而活性下降。
3.2. 生成配体-蛋白复合物相互作用的化学键分析
分子对接后生成配体-蛋白复合物之间相互作用的作用力。PBP2a中的氨基酸残基形成的活性口袋与各个化合物对接时形成的化学键作用力。万古霉素与PBP2a的氨基酸残基ValB 277、LysB 273、AspB:295、AspB 275有氢键作用,与SerB72、AsnB 306等氨基酸存在的范德华力作用,以及与TrpB 205有一个δ-P键的相互作用;对比化合物1,它与PBP2a的结合中形成了以ValB 277、LysB 316、GluB 294有氢键作用,Leu B147存在的范德华力作用和与LysB 273的P-л共轭作用;化合物2与PBP2a的结合中形成了与化合物1相同的LysB 273的P-л共轭作用力,为E-低聚白藜芦醇化合物生成的配体-蛋白复合物相互作用的特征化学键型,以及以ArgA 298、ThrA 08、TyrB 105的氢键作用和SerA 306的范德华力作用力。综上总结不同类别的化合物化学键力特征,万古霉素显然主要是以多药效团的氢键和范德华疏水键力的作用,为主要化学键作用形式;E-低聚白藜芦醇化合物则是以LysB 273的P-л共轭以及在氨基酸残基的活性口袋的分子氢键为主要化学键力的结合方式。总之基于分子对接的化学键的作用和构效关系分析,基本阐明了E-低聚白藜芦醇化合物抑菌杀菌作用显著的机理,与其直接和PBP2a蛋白受体结合有关。
4. 结论
研究证明,万古霉素的作用机制是通过抑制革兰氏阳性菌的细胞壁的合成而使细菌无法生存,阻止细菌繁殖,因而对革兰氏阳性菌具有较好的抑制作用 [14] 。上述分子对接和构效关系的研究可知,万古霉素中大量的亲水基团可以与靶蛋白PBP2a活性中心氨基酸残基形成氢键的相互作用,据报道是与NAM-肽和NAG-肽中D-丙氨酰丙氨酸末端的部分相互作用,这样万古霉素分子就被氢键“捆绑”在了D-丙氨酰丙氨酸上,阻止了NAM-肽与NAG-肽参与肽聚糖骨架的形成。另一方面,已知万古霉素提供了次级结合蛋白信息PBP2a的抗MRSA的作用机制,可有效预测E-低聚白藜芦醇化合物1和2与相关的潜在蛋白PBP2a作用的构效和安全性评价。本文基于我们前期筛选发现的E-低聚白藜芦醇天然化合物1和2的具有的强的抑菌杀菌活性,构效关系研究通过反向分子对接证实的E-低聚白藜芦醇天然化合物1和2,可以表明该类化合物具有类似万古霉素的抗菌机理;然而基于MRSA对甲氧西林的耐药性是有PBP2a所介导的机制,进一步推测E-低聚白藜芦醇天然化合物1和2不同结构作用效能,和对接显示的与靶蛋白PBP2a的相互作用的强的亲和力,有可能具有克服现有β-内酰胺类抗生素存在的耐药性的巨大潜能,或成为克服β-内酰胺类抗生素耐药性的新的药物先导化合物。因为随着对万古霉素耐药性研究的急迫性,寻找抑制高水平PBP2a转肽酶的活性位点小分子探针新颖结构,是克服β-内酰胺类抗生素耐药性所必需的。
基金项目
国家自然科学基金资助项目(No. 21472035)。
NOTES
*通讯作者。