1. 引言
随着纳米技术的飞速发展,越来越多的纳米产品进入人们的生产、生活中,纳米材料已经成为人们研究的热点、重点之一。现有研究范围涉足纳米团簇、纳米线以及纳米薄膜等各个维度,团簇是多个原子聚集而成的凝聚态物质,是构成纳米线、纳米薄膜以及块体材料的基本单元,其研究对纳米材料有重要意义 [1] [2] [3] 。而金属团簇具备奇异的物理、化学性质,在电子技术、高效催化、特殊焊接等领域有重要作用。双金属团簇受异质组元协同效应影响,具有单质团簇没有的特性,但合金团簇的性质受尺寸、异组分、结构和原子分布影响显著 [4] 。因此,人们对双金属团簇的结构与性质开展了深入研究,在贵金属团簇的研究中发现了壳层、孪生、双面特性等特殊结构。Wang Q等人在(AgCo)201团簇的结构演变研究中发现原子亚表面层偏析诱导出Ag-Co-Ag壳层结构 [5] ,在Co-Cu团簇凝固过程中发现fcc与hcp共存孪生结构 [6] ;Kim D H等人在AgPd团簇的固液转变研究中发现了双面特性结构 [7] ;对金、铜、镍等团簇的研究中发现了二十面体结构,团簇在升温过程中出现二十面体转变行为。结构是影响团簇性质的主要因素,我们在Ag-Pd团簇的研究中发现了原子偏析诱导团簇异常熔化 [8] [9] [10] 。因此,本文研究掺杂对Ag团簇结构及相变行为的影响。
2. 基本理论与模拟计算
基本理论:分子动力学方法于1957年由Alder提出 [11] ,后经Rahman、Less、Andersen、Nose等人逐步发展完善,现已成为纳米材料的主要研究手段之一。原子间的相互作用势函数是分子动力学理论的核心,目前已构建了间断势、L-J势以及多体势等。本文选择适用于贵金属材料的嵌入原子势进行模拟计算,原子间的相互作用势采用Zhou等人给出的形式 [12] [13] ,团簇总能量计算公式如下:
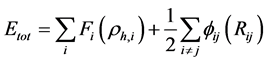 (1)
(1)
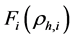 为嵌入项,表示原子
为嵌入项,表示原子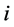 嵌入到团簇内部时产生的嵌入能,
嵌入到团簇内部时产生的嵌入能,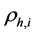 是由除原子
是由除原子 外的所有原子在原子
外的所有原子在原子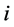 位置处产生的电子密度,
位置处产生的电子密度,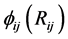 是原子
是原子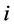 和原子
和原子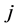 之间的对势,
之间的对势,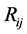 为两原子间的距离。
为两原子间的距离。
模型构建:在尺寸为 的面心立方体结构Ag晶体中心截取外形为截断八面体的Ag团簇,截取原子数量分别为147、309、561个,定义模型中心13个原子为第1层(核心层),依次向外各层原子数为42、92、162、252,截取的团簇模型在300 K弛豫后作为初始模型,弛豫过程共运行50万步,时间步长为1.0 fs。CoAg团簇模型则采用Co原子替换Ag原子,并对构建的合金团簇模型进行弛豫。
的面心立方体结构Ag晶体中心截取外形为截断八面体的Ag团簇,截取原子数量分别为147、309、561个,定义模型中心13个原子为第1层(核心层),依次向外各层原子数为42、92、162、252,截取的团簇模型在300 K弛豫后作为初始模型,弛豫过程共运行50万步,时间步长为1.0 fs。CoAg团簇模型则采用Co原子替换Ag原子,并对构建的合金团簇模型进行弛豫。
模拟计算:热力学升温过程模拟采用正则系综(NVT)运行220万步,时间步长仍然为1.0 fs,模拟过程包含预热、升温、热平衡三个阶段。首先在300 K预热20万步,然后升温到1200 K,共运行180万步,最后在1200 K热平衡20万步,每1000步输出一次运行结果。
3. 计算结果与分析
众所周知,势能是判断材料稳定性的重要参数,材料结构对势能有重要影响,通过势能曲线可以分析材料结构、熔点、热容量等与材料特性息息相关的物理量。现有研究结果表明材料在升、降温过程中,当平均原子势能曲线出现很大跃变时,则出现材料由固体转变为液态的一级相变;当平均原子能曲线无明显跃时,而曲线可以分为斜率不同的两段,即两段曲线的,则体系出现液态与固体的相变,称为二级相变,此时固态为无序结构。本文为了分析团簇升温过程中的结构变化规律,采用平均原子势能随温度变化曲线作为首要判别方式。图1描述了不同尺寸Ag团簇升温过程中平均原子势能随温度变化曲线,原子数分别为147、309和561,团簇原子分布分别为3层、4层、5层。
从图中我们能清楚地看出,三个不同尺寸团簇的势能曲线之间存在明显差异。首先,相同温度下的原子平均势能大小有很大差异,平均原子势能随团簇原子层数减少而增高,我们认为这主要是由于团簇的高表面能和大表体比引起。其次,势能曲线随温度升高的变化有很大差异。Ag309、Ag561团簇的势能曲线在升温过程中出现了大幅度的向上跃变,此时团簇熔化,其对应的熔点分别在758K,837K附近。在团簇熔化前,Ag309、Ag561团簇的势能曲线出现了明显的向下跃变过程,其跃变点温度分别在360K和622K附近,势能曲线的异常表明团簇出现了结构变化。对比Ag309、Ag561团簇的势能曲线,Ag147团簇的势能曲线出现了异常,既没有出现熔点处的向上跃变,也没出现熔化前的向下跃变。
我们通过团簇的外形和原子分布投影图分析Ag147团簇势能曲线的异常以及Ag309、Ag561团簇熔化前的结构变化。图2分别给出了3个团簇的初始结构、升温前后的结构(300 K和1200 K)和升温过程中势能曲线异常变化处的团簇结构外形图以及相对应的原子分布投影图。从图中可以看出在300 K弛豫时不同尺寸的团簇结构发生很大变化,尺寸较大的Ag309、Ag561团簇的结构为面心立方晶格的截断八面体,而尺寸较小的Ag147团簇的结构转变为二十面体。我们借助团簇快照图进一步分析Ag309、Ag561势能曲线在熔化前的异常变化,从快照图可发现两个团簇在升温过程中均出现二十面体结构转变,转变温度分别为360 K和622 K,Ag309团簇的转变温度低,Ag561团簇的转变温度高。由此可分析出二十面体结构的Ag团簇势能低于截断八面体结构的势能,二十面体结构具有稳定性更好,从截断八面体结构转变为二十面体结构的温度随团簇尺寸增大而升高。我们认为团簇发生整体结构转变是多原子体系协同运动的结果,而原子数越多所需要的能量越大,所以转变温度越高。
从升温结束后1200 K的快照图可看出3个团簇均为熔融态,但Ag147团簇势能曲线出现异常变化,升温过程中无明显向上跃变,该团簇在熔化过程中发生二级相变。由于该团簇尺寸小,表面原子比重大,低表面能原子随温度升高出现无序分布,导致团簇在固态时的无序度与熔融态相近,引起势能曲线在团簇熔化时异常。
对比分析三个团簇的模拟结果,我们选择Ag561团簇作为掺杂团簇的基体,其原因主要为Ag561团簇的熔点较高,二十面体结构转变温度较高,团簇为截断八面体固相和二十面体固相的温度范围均较大,通过掺杂调控团簇结构变化范围较宽,有利于增强团簇的应用前景。我们对Ag561团簇进行掺杂研究,掺入原子为磁性材料Co原子,掺入方式采用原子分层替换法,替换原子数量分别为13、55,替换位置从内向外分层进行。
图3为不同初始结构的Co13Ag548团簇升温过程中平均原子势能随温度升高的变化曲线,13个Co从
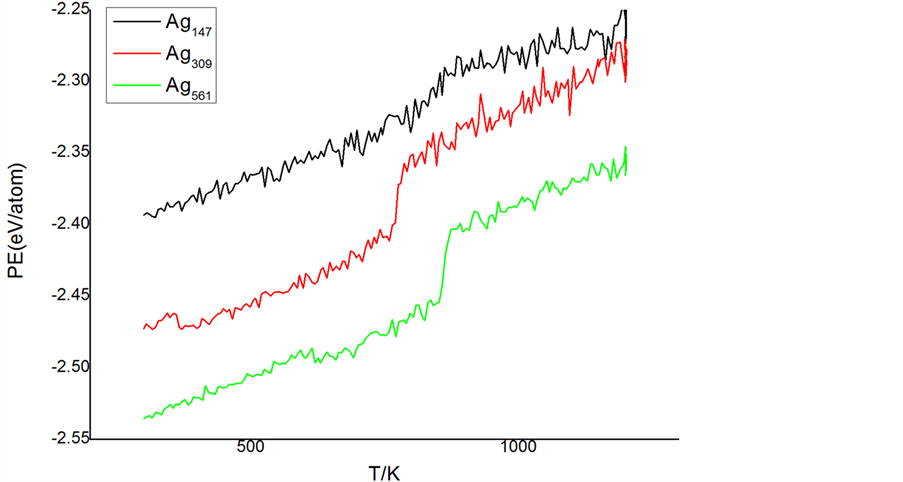
Figure 1. The potential energy curve of Ag clusters with temperature
图1. Ag团簇平均原子势能随温度变化曲线








Figure 2. The snapshot and projection Ag clusters during heating, (a) Ag147, (b) Ag309 and (c) Ag561
图2. Ag团簇升温过程中的外形图与投影,(a) Ag147、(b) Ag309、(c) Ag561
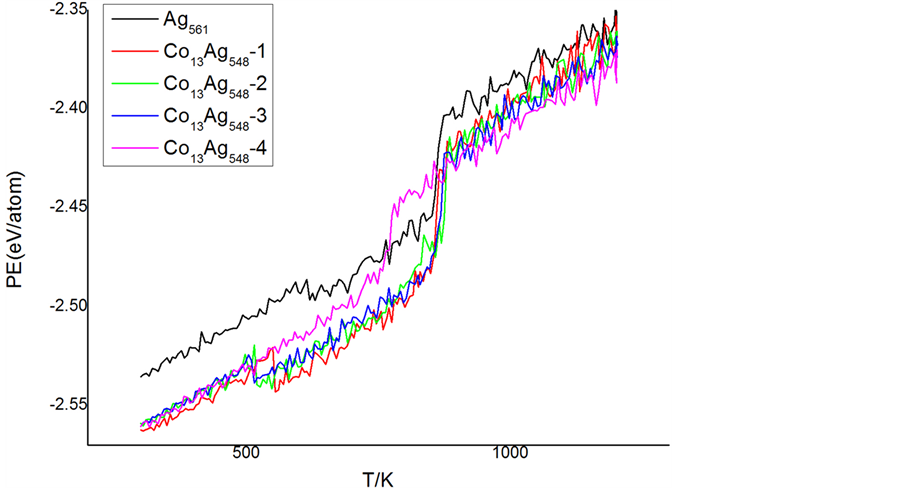
Figure 3. The potential energy curve of Co13Ag548 clusters with temperature
图3. 掺杂团簇Co13Ag548平均原子势能随温度变化曲线
内向外分别替换Ag561团簇1~4层的原子。势能曲线表明掺入Co原子引起团簇势能显著降低,且对团簇二十面体结构转变温度和熔点都有不同影响。在二十面体结构转变方面:Co原子掺入1~3层时,平均原子均势能曲线出现向下跃变,说明团簇出现二十面体结构转变,且转变温度降低,分别在551 K、514 K、502 K附近,而Co原子掺入第4层时,平均原子势能曲线未出现向下跃变,说明团簇升温过程中未出现构转变。在熔点方面,1~3层掺杂Co13Ag548团簇的熔点与Ag561团簇的熔点接近,而掺入第4层时团簇熔点明显降低,在744 K附近。
为此,我们通过原子分布投影的方式进一步对比分析Co原子掺入第1层和第4层时的团簇升温过程中的结构演变,图4所示:(a) 为掺杂第1层;(b) 为掺杂第4层。对比两个团簇快照图可明显看出结构
Figure 4. Snapshots of Co13Ag548 clusters during heating processes, (a) doping Co atoms in first layer, (b) doping Co atoms in fourth layer
图4. Co13Ag548团簇升温过程中结构快照图,(a) Co原子掺入第1层;(b) Co原子掺入第4层
演变过程及其差异,中心掺杂团簇在530 K附件时仍然保持完整的截断八面体结构,Co原子分布在中心位置,温度在551 K附件时,团簇结构转变为二十面体,Co原子位置未发生任何变化,在1200 K时,团簇为熔融态,Co原子分布在第4层。而Co原子掺杂在第4层时,团簇结构演变呈现出异常,从外形图中可看出,团簇在300 K时保持fcc晶格的截断八面体,温度在670 K时,团簇表面层出现局部无序分布,随温度逐步升高,表面层原子无序分布进一步明显,温度为740 K时,团簇表面层原子全部为无序状态,与熔融态团簇表面层原子分布相近,而势能曲线表明团簇未熔化。原子分布投影图给出了合理的解释,团簇温度在670 K时,投影图中的团簇以fcc晶格结构的截断八面体为主,但在表面局部地方出现了无序分布,温度升高到740 K时,团簇内部仍然保持较好的fcc晶格结构,而温度升到780 K时,团簇的fcc晶格结构被完全破坏,团簇熔化。我们认为13个Co原子占第4层原子总数比例太少,随机掺杂导致局部出现多个Co原子聚集现象,由Co原子与Ag原子的势能和性质差异导致升温过程中团出在表面产生局部预熔现象,影响团簇二十面体结构转变,最终引起团簇熔点降低,由此说明二十面体团簇结构转变对熔点有较好的稳定作用。
为了分析掺杂Co原子数量对团簇结构和熔化的影响,我们采用相同的掺杂方式进一步分析Co55Ag506团簇模型,由于掺入原子数增多,将前面的1、2层合并为核心层,即第1层,因此掺杂位置从4层减少为3层。从图5的势能曲线可看出,掺入Co原子数增加引势能曲线异常变化。团簇在升温过程中未发生势能曲线向下跃变,表明团簇熔化前的升温过程中未出现结构转变。中心掺杂的平均原子势能明显低于掺入第2和第3层团簇的势能,表明中心掺杂具有较好的稳定性。中心掺杂团簇Co55Ag506的熔点高于Ag561团簇的熔点,而第2~3层掺杂Co55Ag506的熔点低于Ag561团簇的熔点,与Co13Ag548团簇熔点出现较大差异。
Figure 5. The potential energy curve of Co55Ag506 clusters with temperature
图5. Co55Ag506团簇平均原子势能随温度变化曲线
由于Co55Ag506团簇的势能曲线在固相升温过程中与第4层掺杂的Co13Ag548团簇的势能曲线相似,我们进一步分析Co55Ag506团簇的结构演变。分析发现掺杂在第1和第2层时,团簇在300 K弛豫过程中结构转变为二十面体,掺杂在第3层的二十面体结构转变温度在490 K附近,与Co55Ag506-3势能曲线在490K附近缓慢下降相符合。对于中心掺杂团簇熔点出现升高的异常变化,我们认为是团簇中心的55个Co原子构成稳定性较高的二十面体,与外层Ag原子相对独立的Co核能更好的稳定团簇结构。
4. 结论
我们采用分子动力学方法模拟研究了Ag561及其掺杂团簇的升温过程和团簇结构演变过程,采用平均原子势能曲线和结构快照图表示各团簇结构转变和熔点。研究发现Co掺杂对Ag561团簇升温过程中结构演变有重要作用,掺入Co原子引起团簇二十面体结构转变温度降低,在第4层掺入13个Co原子时未发现二十面体结构转变异常变化,在相同位置掺入55个Co原子的二十面体结构转变温度低于13个Co原子时的结构转变温度。掺杂Co原子对团簇平均原子势能有作用,掺入Co原子数越多团簇势能越低,掺入相同原子数时中心掺杂团簇势能最低。掺杂对团簇熔点也有重要影响,在1~3掺入13个Co原子的团簇受二十面体结构作用熔点未发生明显变化,在第4层掺杂时受Co原子预熔作用的影响,未发生二十面体结构转变导致熔点显著降低的异常,而掺入55个Co原子时团簇均出现二十面体结构转变,但中心掺杂引起团簇熔点升高,2~3层掺杂引起熔点降低。
基金项目
重庆市教委科学技术研究项目(批准号:KJ1401108);重庆市高校微纳米材料工程与技术重点实验室开放课题(批准号:KFJJ1404);重庆文理学院项目(批准号:Y2015CH33)。